Volume 10, Number 7—July 2004
Dispatch
Phylogenetic Analysis of West Nile Virus, Nuevo Leon State, Mexico
Figure 1
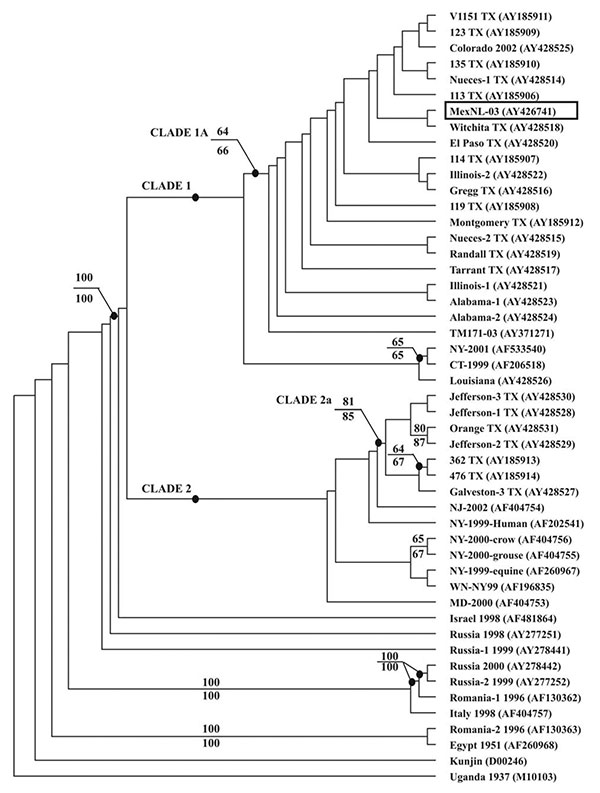
Figure 1. Phylogenetic analysis of West Nile virus (WNV) from Nuevo Leon State, Mexico. Phylogenies were estimated using the program MRBAYES, version 2.0 (15). Sampling of trees from the posterior probability distribution used the Metropolis-coupled Markov chain Monte Carlo algorithm to allow running of multiple Markov chains. A run with four chains was performed for 90,000 generations, under a general time-reversible model (all six types of substitutions occur at different rates) with parameter value estimation for base frequencies, substitution matrix values, and rate heterogeneity. Rate heterogeneity was estimated using a γ distribution for the variable sites and assuming a certain portion of sites to be invariable. The burn-in time was 70,000 generations. The phylogenetic analysis is based on the 2004-nt fragment encoding the complete prM-E genes of 49 WNVs. The tree is rooted by using the prototype WNV strain from Uganda in 1937 (GenBank accession no. M10103) as an outgroup. Values above some branches represent the percent support by parsimony bootstrap analysis. Values below some branches represent the percentage support by distance bootstrap analysis. The bootstrap confidence estimates are based on 1,000 replicates. The WNV from Nuevo Leon State is encapsulated.
References
- Nash D, Mostashari F, Fine A, Miller J, O’Leary D, Murray K, The outbreak of West Nile virus infection in the New York City area in 1999. N Engl J Med. 2001;344:1807–14. DOIPubMedGoogle Scholar
- Centers for Disease Control and Prevention. West Nile virus—statistics, surveillance and control, September 30, 2003. [cited 2004 May 14]. Available from: http://www.cdc.gov/ncidod/dvbid/westnile/surv&control.htm
- Health Canada. Population and Public Health Branch WNV surveillance updates, September 30, 2003. [cited 2004 May 14]. Available from: http://www.hc-sc.gc.ca/pphb-dgspsp/wnv-vwn/mon_e.html#sitrep.
- Blitvich BJ, Fernández-Salas I, Contreras-Cordero JF, Marlenee NL, González-Rojas JI, Komar N, Serologic evidence of West Nile virus infection in horses, Coahuila State, Mexico. Emerg Infect Dis. 2003;9:853–6.PubMedGoogle Scholar
- Estrada-Franco JG, Navarro-Lopez R, Beasley DWC, Coffey L, Carrara A-S, Travassos da Rosa A, West Nile virus in Mexico: serologic evidence of widespread circulation since July 2002. Emerg Infect Dis. 2003;9:1604–7.PubMedGoogle Scholar
- Loroño-Pino MA, Blitvich BJ, Farfán-Ale JA, Puerto FI, Blanco JM, Marlenee NL, Serologic evidence for West Nile virus infection in horses, Yucatán State, México. Emerg Infect Dis. 2003;9:857–9.PubMedGoogle Scholar
- Ulloa A, Langevin SA, Mendez-Sanchez JD, Arredondo-Jimenez JI, Raetz JL, Powers AM, Serologic survey of domestic animals for zoonotic arbovirus infections in the Lacandón Forest region of Chiapas, Mexico. Vector Borne Zoonotic Dis. 2003;3:3–9. DOIPubMedGoogle Scholar
- Lanciotti RS, Roehrig JT, Deubel V, Smith J, Parker M, Steele K, Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science. 1999;286:2333–7. DOIPubMedGoogle Scholar
- Lanciotti RS, Ebel GD, Deubel V, Kerst AJ, Murri S, Meyer R, Complete genome sequences and phylogenetic analysis of West Nile virus strains isolated from the United States, Europe, and the Middle East. Virology. 2002;298:96–105. DOIPubMedGoogle Scholar
- Briese T, Glass WG, Lipkin WI. Detection of West Nile virus sequences in cerebrospinal fluid. Lancet. 2000;355:1614–5. DOIPubMedGoogle Scholar
- Anderson JF, Vossbrinck CR, Andreadis TG, Iton A, Beckwith WH, Mayo DR. A phylogenetic approach to following West Nile virus in Connecticut. Proc Natl Acad Sci U S A. 2001;98:12885–9. DOIPubMedGoogle Scholar
- Ebel GD, Dupuis AP, Ngo K, Nicholas D, Kauffman E, Jones SA, Partial genetic characterization of West Nile virus strains, New York State, 2000. Emerg Infect Dis. 2001;7:650–3. DOIPubMedGoogle Scholar
- Beasley DW, Davis CT, Guzman H, Vanlandingham DL, Travassos da Rosa AP, Parsons RE, Limited evolution of West Nile virus has occurred during its southwesterly spread in the United States. Virology. 2003;309:190–5. DOIPubMedGoogle Scholar
- Davis TC, Beasley DWC, Guzman H, Raj P, D’Anton M, Novak RJ, Genetic variation among temporally and geographically distinct West Nile virus isolates collected in the United States during 2001 and 2002. Emerg Infect Dis. 2003;9:1423–9.PubMedGoogle Scholar
- Huelsenbeck JP, Ronquist FR. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–5. DOIPubMedGoogle Scholar
- Rappole JH, Hubalek Z. Migratory birds and West Nile virus. J Appl Microbiol. 2003;94(Suppl):47S–58S. DOIPubMedGoogle Scholar
- Fernández-Salas I, Contreras-Cordero JF, Blitvich BJ, González-Rojas JI, Cavazos-Alvarez A, Marlenee NL, Serologic evidence of West Nile virus infection in birds, Tamaulipas State, México. Vector Borne Zoonotic Dis. 2003;3:209–13. DOIPubMedGoogle Scholar
- Farfán-Ale JA, Blitvich BJ, Loroño-Pino MA, Marlenee NL, Rosado-Paredes EP, García-Rejón JE, Longitudinal studies of West Nile virus infection in avians, Yucatán State, México. Vector Borne Zoonotic Dis. 2004;4:3–14. DOIPubMedGoogle Scholar