Volume 11, Number 1—January 2005
Research
Hybrid Vibrio vulnificus
Abstract
The recent emergence of the human-pathogenic Vibrio vulnificus in Israel was investigated by using multilocus genotype data and modern molecular evolutionary analysis tools. We show that this pathogen is a hybrid organism that evolved by the hybridization of the genomes from 2 distinct and independent populations. These findings provide clear evidence of how hybridization between 2 existing and nonpathogenic forms has apparently led to the emergence of an epidemic infectious disease caused by this pathogenic variant. This novel observation shows yet another way in which epidemic organisms arise.
Vibrio vulnificus, a ubiquitous inhabitant of marine and estuarine environments, is considered one of the most dangerous waterborne pathogens. The case-fatality rate for V. vulnificus septicemia may reach 50% (1). Human infection is generally acquired through eating contaminated raw or undercooked seafood or through contamination of wounds by seawater or marine animals (2). Infected persons with preexisting liver disease, hemochromatosis, or compromised immune systems are at particularly high risk for fatal septicemia (3–8).
Human infections are sporadic and almost entirely caused by strains of biotype 1, while biotype 2 strains have been reported to cause disease mainly among eels and rarely infect humans (9). During the summer of 1996, a major outbreak of systemic V. vulnificus infections started among Israeli fish market workers and fish consumers (10,11). Molecular studies showed that the disease outbreak was caused by a previously undescribed biotype that exhibited a distinct phenotypic and molecular pattern, designated biotype 3 (11).
The origins of this emergent infectious disease have not been fully understood, although it was originally thought to arise mostly from human behavior and work practices (10). On the basis of these assumptions, new fish-handling procedures were introduced (11,12). However, disease continued, although at a lower incidence. Therefore, studies were undertaken to determine whether this novel outbreak of disease was caused by a specific lineage or clone. The emergence of this new biotype could not be resolved by conventional microbiologic and molecular typing approaches. We investigated this outbreak by combining a multilocus sequence typing approach (13) with molecular evolutionary analyses.
Bacterial Isolates
To study the emergence of this new biotype, we examined a collection of 159 V. vulnificus isolates that represented all 3 biotypes from human disease and environmental sources that originated in Israel (n = 64), the Untied States (n = 54), Denmark (n = 7), Germany (n = 6), Spain (n = 5), Sweden (n = 5), Japan (n = 8), South Korea (n = 2), Singapore (n = 2), Thailand (n = 1), Indonesia (n = 1), and Taiwan (n = 1). In addition, 3 well-characterized reference strains that represented the 3 biotypes were included, the ATCC 27562 strain (biotype 1, isolated from human blood in the USA), the E-39 strain (biotype 2, isolated from diseased eel in Spain), and ATCC BAA-86 (biotype 3, isolated from human blood in Israel). Biotype 1 strains (n = 82) consisted of 39 isolates from human disease and 43 environmental isolates; biotype 2 strains (n = 15) consisted of 13 isolates from diseased eels, 1 from and infected person, and 1 from diseased shrimp. Biotype 3 strains were isolated from samples from persons with invasive disease in Israel (n = 61) and from fish-pond water (n = 3). Isolates were grown on blood agar plates and incubated overnight at 35°C in aerobic conditions. The lists of the isolates used in this study and their sources can be accessed at http://pubmlst.org/vvulnificus.
DNA Extraction
The DNeasy kit (QIAGEN GmbH, Hilden, Germany) was used to extract DNA with the gram-negative bacterial protocol as recommended by the manufacturer. Briefly, several colonies from a bacterial culture were picked off into phosphate-buffered saline solution and centrifuged at 7,500 rpm (5,000 x g) for 10 min. The cell pellet was resuspended in 180 μL of tissue lysis buffer, then 20 μL of proteinase K (10 mg/mL) was added, and the sample was incubated at 55°C until the tissue was completely lysed. Then 200 μL of lysis buffer was added and incubated at 70°C for 10 min. The DNA in the clear viscous lysates was precipitated with ethanol 95% (vol/vol) and added to DNeasy mini columns. Ethanol 70% (vol/vol)–based buffers (AW1 and AW2) were added sequentially to the columns and centrifuged at 8,000 rpm (6,000 x g). The supernatants were discarded, and the DNA was resuspended in AE buffer and used for amplification.
Multilocus Sequence Typing (MLST)
This bacterium has 2 chromosomes. Fourteen housekeeping genes (7 from each chromosome) that encoded enzymes responsible for intermediary metabolism were identified by searching the genome database (http://www.ncbi.nlm.nih.gov/genomes/MICROBES/Complete.html) of V. vulnificus strain CMCP6, with gene sequences from other bacteria. Genetic loci were chosen for further investigation on the basis of the following criteria: chromosomal location, suitability for primer design, and sequence diversity in pilot studies. Ten loci were chosen for the MLST scheme, 5 from each chromosome. The following were chosen from the large chromosome: glp, the encoding glucose-6-phosphate isomerase; gyrB, the encoding DNA gyrase-subunit B; mdh, the encoding malate-lactate dehydrogenase; metG, the encoding methionyl-tRNA synthetase; and purM, the encoding phosphoribosylaminoimidazole synthetase. The following were chosen from the small chromosome: dtdS, the encoding threonine dehydrogenase; lysA, the encoding diaminopimelate decarboxylase; pntA, the encoding transhydrogenase alpha subunit;; pyrC, the encoding dihydroorotase; and tnaA, the encoding tryptophanase. Their chromosomal location suggested that it was unlikely for any of the loci to be coinherited in the same recombination event, as the minimum distance between loci was 300 kb (Table).
Amplification and Nucleotide Sequence Determination
Polymerase chain reaction (PCR) products were amplified with oligonucleotide primer pairs designed from the V. vulnificus genome sequence. These primers provided reliable amplification from a diverse range of samples (available from http://pubmlst.org/vvulnificus). Each 50-μL amplification reaction mixture was made up of 10 ng of V. vulnificus chromosomal DNA, 100 pmol of each PCR primer (MWG Biotech, Ebersberg, Germany), 10 x PCR buffer with 1.5 mM MgCl2 (QIAGEN GmbH), 0.5 U of Taq DNA polymerase (QIAGEN GmbH), and 1.6 mM deoxynucleoside triphosphates (ABgene, Epsom, UK). The reaction conditions were denaturation at 94°C for 1 min, primer annealing at 50°C for 45 s and extension at 72°C for 1 min for 30 cycles. The amplification products were purified by precipitation with 20% polyethylene glycol and 2.5 M NaCl (14), and their nucleotide sequences were determined at least once on each DNA strand by using internal nested primers (available from http://pubmlst.org/vvulnificus) and ABI PRISM BigDye Terminators v 3.0 Reaction Mix (Applied Biosystems, Foster City, CA) in accordance with the manufacturer’s instructions. Unincorporated dye terminators were removed by precipitation of the termination products with sodium acetate (3 M, pH 5.2) and 95% ethanol, and the reaction products were separated and detected with an ABI PRISM 3730 DNA Analyzer (Applied Biosystems). Sequences were assembled from the resultant chromatograms with the STADEN suite of computer programs and edited to resolve any ambiguities (15). For each locus, every different sequence was assigned a distinct allele number in order of identification; these sequences were internal fragments of the gene, which contained an exact number of codons. Each isolate was therefore designated by a 10-integer number (the allelic profile), which corresponds to the allele numbers at the 10 loci in the following order: glp, gyrB, mdh, metG, purM, dtdS, lysA, pntA, pyrC, and tnaA. Isolates with the same allelic profile are assigned to the same sequence type (ST), which were numbered in the order of their identification (ST-1, ST-2, and so on). The data have been deposited (http://pubmlst.org/vvulnificus).
Inferring the Population Structure and Ancestral Sources
The program STRUCTURE was used to define the population structure and identify the ancestral sources of the 10 gene fragments from all the strains. STRUCTURE is a recently developed program that implements a Bayesian model approach for inferring population structure and ancestral sources from multilocus genotype data (16). Of the 4,326 nucleotides sequenced for each isolate from the 10 genes, 447 nt were polymorphic. For the purposes of the analysis, these nucleotides were used by STRUCTURE as individual loci. STRUCTURE can infer the population structure by using a variety of models, including the linkage model (17), which incorporates linkage disequilibrium due to correlations in ancestry between loci that reflects admixture between populations. This approach has recently been used to elucidate the structure and evolution of populations of the human pathogen Helicobacter pylori (18), and we have used the same method here. For the purposes of the analysis, the nucleotide sequence of the 10 housekeeping gene fragments of 2 clinical strains of V. vulnificus, CMCP6 and YJ016, whose complete genome sequence has been recently completed (http://www.ncbi.nlm.nih.gov/genomes/MICROBES/Complete.html), were added to all the data sets.
Genotypes Identified
The 159 isolates were resolved into 70 STs, 56 of which were present only once in the entire collection. Eighty-two isolates (51.6%) were represented by 1 of 4 STs: ST-8 was the most common and occurred 62 times (39%); ST-6 occurred in 11 isolates (6.9%); ST-32 occurred in 5 isolates (3.1%); ST-16 occurred in 4 isolates (2.5%). The remaining 21 isolates resolved into 10 sequence types. Strains of biotype 1 (n = 82) resolved into 66 STs. Biotype 2 (15 isolates) resolved into 4 STs; ST-6, ST-9, ST-10, and ST-48. ST-6 was the most common, occurring 11 times and consisting of all the indole-negative isolates. All biotype 3 strains (n = 62) were genetically identical and belonged to ST-8.
Population Structure and Ancestral Sources
Figure 1
Figure 1. Triangle plots of STRUCTURE results. Only 2 populations were identified (A and B). shows the distribution of the biotypes within the 2 populations. B shows the distribution of the strains...
The observed sequence variation between the 2 chromosomes was comparable. Initial analysis of sequence data from the 10 gene fragments showed that extensive recombination had occurred within all the genetic loci under study (data not shown). Constructing phylogenetic trees in the presence of recombination is problematic because different parts of the sequence may have different phylogenetic histories. Therefore, we analyzed the data with the program STRUCTURE. First, we tested the assumption that the 3 V. vulnificus biotypes represent 3 distinct predetermined populations of this pathogen (K = 3). The results of multiple analyses with STRUCTURE were incompatible with this assumption; in all cases, only 2 populations were identified, populations A and B (Figure 1 and data not shown). Further, while biotype 1 was present in both populations, biotype 2 was present only in population A (Figure 1A). Biotype 3 occupied an intermediate position between the 2 populations (Figure 1A). Figure 1B shows that an overrepresentation of human disease isolates occurred in population B and an overrepresentation of environmental isolates occurred in population A. And Figure 1C shows that both populations were globally distributed.
Figure 2
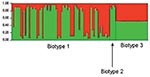
Figure 2. Results of a Bayesian cluster analysis by STRUCTURE. Each of the strains included in the analysis is represented by a thin vertical line, partitioned into 2 colored segments that represent the...
Figure 3

Figure 3. A neighbor-joining tree of representative isolates from the 2 populations is plotted with the inferred ancestral sources of individual polymorphic sites in each of the 10 genes. The names of the...
To identify the evolutionary processes underlying the emergence of the genotype responsible for the Israeli outbreak, we repeated the STRUCTURE analysis assuming only 2 populations (K = 2) (based on the findings from the first STRUCTURE analysis). This analysis identified the ancestral sources of the individual strains (Figure 2). Each strain is represented by a thin vertical line partitioned into 2 (K = 2) most likely predetermined populations or genetic ancestries. Each line shows the proportion of polymorphic sites inherited from each of the 2 populations (shown in green and red). It shows that most biotype 1 and 2 strains have predominant contribution from 1 of the 2 genetic ancestries. However, strains of biotype 3 have almost equal contributions from both genetic ancestries. This analysis was further detailed to identify the ancestral sources of each of the polymorphic sites in each of the 10 gene fragments (Figure 3).These analyses confirmed that, notwithstanding the subdivision of V. vulnificus populations into 2 populations, recombination had occurred between these populations and that the Israeli outbreak genotype is a hybrid, with some genes originating from 1 population and some from another, while some genes have representation from both.
We have shown that a hybrid virulent organism that acquired genes from 2 distinct and independent populations has caused the disease outbreak in Israel. To achieve this analysis, we studied large, carefully assembled, collections of V. vulnificus isolates. The human strains were collected from infected patients in Israel, the United States, Europe, and Southeast Asia. The environmental strains were collected from environmental sources in the United States, the Pacific Ocean, the Baltic Sea, inland fish farms in Israel, and eel farms in Europe.
The division of V. vulnificus populations into 2 major groups is consistent with results of multilocus enzyme electrophoresis studies (19). However, those studies placed the Israeli electrophoretic type within 1 of the 2 groups, in contrast to our findings, which placed the Israeli genotype in an intermediate position between the 2 populations (Figure 1A and Figure 3).
Hybridization within bacterial populations, i.e., the process whereby a hybrid results from the hybridization of the genomes of 2 or more populations of a species or an organism, has been the focus of much attention by scientists in the last decade (20–25); these events, which may be intra- or interspecies, could alter the genetic distances and the phylogenetic relationships within bacterial populations. The magnitude by which these events occur is crucially dependent on ecologic factors; different populations of a species must be present within the same niche for genetic exchange to have an impact on genetic variation (26). Multiple sampling of fish-farm water and fish documented the abundance of biotype 1 strains (11 and data not shown). These biotype 1 strains, representing both populations of V. vulnificus, were never implicated in disease among fish-farm fish, according to the Central Fish Health Laboratory, Kibbutz Nir David, Israel (www.moag.gov.il/english), or among humans in Israel (11). These observations are consistent with finding that these populations are not pathogenic to either humans or fish. The finding that this hybrid variant (biotype 3) was the only implicated organism in all disease cases from 1995 to 2003 is indicative of its pathogenicity. Furthermore, the finding that all 62 biotype 3 strains were genetically identical could suggest that this hybrid clone may have evolved by a relatively recent genome hybridization event.
Hybrid variants have been recently described among populations of Staphylococcus aureus (27) and Chlamydia trachomatis (28). However, our findings show the first bacterial variant that is clearly more pathogenic than the existing forms of the organism, i.e., the Israeli hybrid clone, is more pathogenic than the existing biotype 1 strains within the Israeli aquaculture system. These findings are consistent with observations among influenza viruses (29). This phenomenon has also recently been described also among populations of mosquitoes (30), in which hybridization between existing forms of a relatively nonpathogenic organism has apparently led to the emergence of a novel pathogenic variant that poses a particular threat to human health.
The Israeli genotype spread extensively after its emergence in 1995, and by 2003, most of the fish farms in Israel were the sources of V. vulnificus cases. This finding is consistent with the idea that this pathogen is circulating freely within the underground brackish water reservoirs that supply these fish farms. Despite the widespread use of inland fish farming around the world, no similar outbreaks have been reported. During the 1970s and 1980s, the introduction into Israel of stocks of Tilapia spp. from Africa, the Far East, and South America (31–33) (for experimental and commercial purposes) may have contributed to the evolution of this hybrid clone. In view of the widespread fish-trading industry, this hybrid clone may eventually emerge through exports of Israeli tilapia stocks, in remote geographic locations. In conclusion, these observations demonstrate the power of molecular and population genetic approaches in investigating the emergence of a novel pathogen and defining its nature. Our results show another way by which epidemic infectious diseases arise.
Dr. Bisharat is infectious diseases physician in Israel currently working as a Wellcome Trust Research Fellow at Oxford University. His main research interests are infectious diseases epidemiology and the emergence of Vibrio vulnificus in Israel.
Acknowledgments
We thank Raul Colodner, Larisa Lerner, Angelo DePaola, James Oliver, Astrid Lewin, Sumio Shinoda, Shin-ichi Miyoshi, Inger Dalsgaard, and Luis Torres for providing bacterial strains and Keith Jolley for providing support in database and Web site maintenance.
This work was supported by the Wellcome Trust, Grant number: 067147/Z/02/Z. Naiel Bisharat is the recipient of a grant from the Wellcome Trust Travelling Research Fellowships. Martin C. Maiden is a Wellcome Trust Senior Research Fellow in basic biomedical sciences.
References
- Centers for Disease Control and Prevention. Vibrio vulnificus infections associated with raw oyster consumption—Florida, 1981–1992. MMWR Morb Mortal Wkly Rep. 1993;42:405–7.PubMedGoogle Scholar
- Hlady WG, Klontz KC. The epidemiology of Vibrio infections in Florida, 1981–1993. J Infect Dis. 1996;173:1176–83. DOIPubMedGoogle Scholar
- Levine WC, Griffin PM. Vibrio infections on the Gulf Coast: results of first year of regional surveillance. Gulf Coast Vibrio Working Group. J Infect Dis. 1993;167:479–83.PubMedGoogle Scholar
- Bullen JJ, Spalding PB, Ward CG, Gutteridge JM. Hemochromatosis, iron and septicemia caused by Vibrio vulnificus. Arch Intern Med. 1991;151:1606–9. DOIPubMedGoogle Scholar
- Laosombat V, Pruekprasert P, Wongchanchailert M. Non-0:1 Vibrio cholerae septicemia in thalassemia patients. Southeast Asian J Trop Med Public Health. 1996;27:411–3.PubMedGoogle Scholar
- Kizer KW. Vibrio vulnificus hazard in patients with liver disease. West J Med. 1994;161:64–5.PubMedGoogle Scholar
- Barton JC, Coghlan ME, Reymann MT, Ozbirn TW, Acton RT. Vibrio vulnificus infection in a hemodialysis patient receiving intravenous iron therapy. Clin Infect Dis. 2003;37:e63–7. DOIPubMedGoogle Scholar
- Katz BZ. Vibrio vulnificus meningitis in a boy with thalassemia after eating raw oysters. Pediatrics. 1988;82:784–6.PubMedGoogle Scholar
- Amaro C, Biosca EG. Vibrio vulnificus biotype 2, pathogenic for eels, is also an opportunistic pathogen for humans. Appl Environ Microbiol. 1996;62:1454–7.PubMedGoogle Scholar
- Bisharat N, Raz R. Vibrio infection in Israel due to changes in fish marketing. Lancet. 1996;348:1585–6. DOIPubMedGoogle Scholar
- Bisharat N, Agmon V, Finkelstein R, Raz R, Ben-Dror G, Lerner L, Clinical, epidemiological, and microbiological features of Vibrio vulnificus biogroup 3 causing outbreaks of wound infection and bacteraemia in Israel. Israel Vibrio Study Group. Lancet. 1999;354:1421–4. DOIPubMedGoogle Scholar
- Bisharat N. Vibrio vulnificus infections can be avoided. Isr Med Assoc J. 2002;4:631–3.PubMedGoogle Scholar
- Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A. 1998;95:3140–5. DOIPubMedGoogle Scholar
- Embley TM. The linear PCR reaction: a simple and robust method for sequencing amplified rRNA genes. Lett Appl Microbiol. 1991;13:171–4. DOIPubMedGoogle Scholar
- Staden R. The staden sequence analysis package. Mol Biotechnol. 1996;5:233–41. DOIPubMedGoogle Scholar
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59.PubMedGoogle Scholar
- Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–87.PubMedGoogle Scholar
- Falush D, Wirth T, Linz B, Pritchard JK, Stephens M, Kidd M, Traces of human migrations in Helicobacter pylori populations. Science. 2003;299:1582–5. DOIPubMedGoogle Scholar
- Gutacker M, Conza N, Benagli C, Pedroli A, Bernasconi MV, Permin L, Population genetics of Vibrio vulnificus: identification of two divisions and a distinct eel-pathogenic clone. Appl Environ Microbiol. 2003;69:3203–12. DOIPubMedGoogle Scholar
- Dykhuizen DE, Green L. Recombination in Escherichia coli and the definition of biological species. J Bacteriol. 1991;173:7257–68.PubMedGoogle Scholar
- Lan R, Reeves PR. Gene transfer is a major factor in bacterial evolution. Mol Biol Evol. 1996;13:47–55.PubMedGoogle Scholar
- Smith JM, Dowson CG, Spratt BG. Localized sex in bacteria. Nature. 1991;349:29–31. DOIPubMedGoogle Scholar
- Lawrence JG, Ochman H. Molecular archaeology of the Escherichia coli genome. Proc Natl Acad Sci U S A. 1998;95:9413–7. DOIPubMedGoogle Scholar
- Li J, Nelson K, McWhorter AC, Whittam TS, Selander RK. Recombinational basis of serovar diversity in Salmonella enterica. Proc Natl Acad Sci U S A. 1994;91:2552–6. DOIPubMedGoogle Scholar
- Martin W. Mosaic bacterial chromosomes: a challenge en route to a tree of genomes. Bioessays. 1999;21:99–104. DOIPubMedGoogle Scholar
- Feil EJ, Spratt BG. Recombination and the population structures of bacterial pathogens. Annu Rev Microbiol. 2001;55:561–90. DOIPubMedGoogle Scholar
- Robinson DA, Enright MC. Evolution of Staphylococcus aureus by large chromosomal replacements. J Bacteriol. 2004;186:1060–4. DOIPubMedGoogle Scholar
- Millman K, Black CM, Johnson RE, Stamm WE, Jones RB, Hook EW, Population-based genetic and evolutionary analysis of Chlamydia trachomatis urogenital strain variation in the United States. J Bacteriol. 2004;186:2457–65. DOIPubMedGoogle Scholar
- Shaw M, Cooper L, Xu X, Thompson W, Krauss S, Guan Y, Molecular changes associated with the transmission of avian influenza a H5N1 and H9N2 viruses to humans. J Med Virol. 2002;66:107–14. DOIPubMedGoogle Scholar
- Fonseca DM, Keyghobadi N, Malcolm CA, Mehmet C, Schaffner F, Mogi M, Emerging vectors in the Culex pipiens complex. Science. 2004;303:1535–8. DOIPubMedGoogle Scholar
- Matty AJ. Aquaculture in Israel. London: Anglo-Israel Association; 1982.
- Hepher B, Pruginin Y. Commercial fish farming. New York: John Wiley & Sons; 1981.
- Sarig S. The development of polyculture in Israel: a model of intensification. In: Shepherd CJ, Bromage N, editors. Intensive fish farming. Oxford: BSP Professional Books; 1988.
Figures
Table
Cite This ArticleTable of Contents – Volume 11, Number 1—January 2005
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Naiel Bisharat, Department of Medicine, Ha’Emek Medical Centre, Afula 18101, Israel; fax: 972-4-6495134
Top