Volume 11, Number 11—November 2005
Dispatch
Vibrio cholerae Pathogenic Clones
Abstract
We resolved the relationships between 2 pandemic clones of Vibrio cholerae. Using 26 housekeeping genes, we showed that the US Gulf clone, the Australian clone, and 3 El Tor strains isolated before the seventh pandemic were related to the seventh pandemic clone. The sixth pandemic clone was well separated from them.
Cholera caused by Vibrio cholerae is a major disease that has caused great fear since the first recorded pandemic in 1817 because of the frequency of death and the rapidity with which it occurs (1,2). Approximately 200 O antigens have been distinguished serologically (3,4), but only O1 and O139 have been found in epidemic and pandemic cholera isolates (5,6).
The seventh pandemic (1961–present) is still widespread and has a severe impact on 3 continents. The sixth pandemic ended in 1923, but the clone persisted at least until the 1990s (7). Furthermore, several cholera outbreaks were reported after the sixth pandemic retreated but before the start of the seventh pandemic. Isolates from these outbreaks were recognized as different from those of the sixth pandemic and were allocated to the El Tor biotype, while the sixth and fifth pandemics, both of which had been studied microbiologically, were referred to as the classical biotype. These El Tor outbreaks occurred in Indonesia and the Middle East (1926–1960) (5) and are often referred to as prepandemic isolates because they were later seen as forerunners of the seventh pandemic, which was also of the El Tor biotype. However, now that environmental V. cholerae has been studied in some detail, major components of the El Tor phenotype are known to be present in most environmental isolates, and the classical biotype is believed to have arisen by loss of characters otherwise widely present in the species (8). Also, cases of sporadic indigenous cholera have been detected in Australia (9) and the United States (10), both of the O1 El Tor biotype. These are generally referred to as the US Gulf and Australian clones. All of the pathogenic forms discussed above had the O1 serotype, but in 1992 a variant of the seventh pandemic appeared with a new O antigen, O139; this variant is known as V. cholerae O139 Bengal (11).
The relationships of V. cholerae have been studied in several ways, but the most useful insights have come from multilocus enzyme electrophoresis (12) and more recently by multilocus sequence analyses (4,13,14). In this study, we sequenced 26 housekeeping genes from V. cholerae isolates representative of the sixth and seventh pandemic clones and other closely related toxigenic strains to determine relationships to better understand the origins of pandemic clones.
Twenty-six housekeeping genes distributed evenly on both chromosomes (Table A1) were studied by using 5 nontoxigenic environmental isolates and 12 toxigenic V. cholerae isolates, which comprise 5 sixth and seventh pandemic isolates and 7 pathogenic isolates related to them (Table). One of the 3 seventh pandemic isolates is N16961; the sequence of its genome (15) was used in this study. All 26 housekeeping genes were successfully sequenced from the remaining 11 toxigenic isolates. However, 4 genes could not be amplified by polymerase chain reaction from 1 of the environmental isolates (sdaA for 370-94, hmpA for 905-93, glgX for 928-93, and pepN for 370-94) and were not sequenced. The GenBank accession numbers for the nucleotide sequences determined in this study are DQ020969–DQ021380.
The 5 nontoxigenic environmental isolates had different sequences for each gene. These sequences were not found in any of the 12 toxigenic isolates. The average pairwise difference among the 5 environmental isolates in the 22 genes sequenced ranged from 0.67% to 5.29%, an indication that V. cholerae as a species has a high level of sequence variation. However 11 of the 26 housekeeping genes (glyA, gppA, pntA, icd, purM, plsX, ndh, glgX, adk, carA, and speA) were identical in all toxigenic V. cholerae isolates, confirming that they are closely related.
Figure 1

Figure 1. Distribution of base differences in strains of Vibrio cholerae. The base positions relative to the start of the ATG codon are shown. Isolates 395 and E9120 were compared for the fruK,...
There were 18 mutation/recombination events among the 15 genes with sequence variation in the 12 toxigenic isolates. Three (malP, pyrC, and gyrB) of these genes had undergone 2 changes. Seven cases of single base differences, which are attributed to mutation, were clearly distinct from 11 cases of multiple base differences, which are attributed to recombination. Ten of the events attributed to recombination involve changes in 10 to 51 bases (Figure 1). The metG gene, in which the 2 sequences differ by only 4 bases, might have undergone 4 mutational events rather than a single recombination event, but we considered this to be most unlikely.
Some of the 11 genes believed to have undergone recombination may have undergone >1 recombination event. This possibility is likely because 11 of 26 genes have undergone recombination. For example, in metE the base changes are at the 2 ends and may represent 2 recombination events; the same applies, to a lesser extent, to malP.
With regard to the length of DNA involved in recombination events, segments longer than a gene are most common because only in the genes discussed above and in sdaA and gyrB are the bases that differed clustered within the gene. This is suggestive of recombination within that gene.
Figure 2
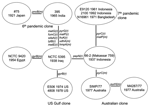
Figure 2. Relationships of toxigenic Vibrio cholerae isolates. The mutational (m) and recombinational (r) changes are given equal weight. Shown is a unique (unrooted) tree, with the events indicated on the branches.
The sequences for the 26 genes were used to produce a tree with mutational and recombinational events and given equal weight, which is shown in Figure 2. By sequencing 26 genes, we have observed sufficient variation to determine the relationships of the isolates studied. The tree is unrooted because the environmental isolates share no alleles with each other or toxigenic isolates. The relatively high rate of recombination in V. cholerae means that the level of sequence similarity does not indicate relatedness of the isolates unless the sequences are very similar and differences can be attributed to mutation, as in the toxigenic strains. However, other reasons exist for believing that the root is on the long 10-event branch that includes 2 mutational changes. The 10 El Tor isolates on 1 end are, at most, 3 steps from it and the 2 classical sixth pandemic isolates are separated by 1 event. The isolates date from 1937 to 1992 for the former group and from 1921 to 1965 for the latter group. Since 1 group giving rise to the other while undergoing little divergence itself is highly improbable, we believe that the root lies somewhere on that branch. The properties that characterize the El Tor biotype are those of environmental strains, which makes it unlikely that they are derived from the sixth pandemic, and the reverse seems even less likely because the fifth pandemic was also the classical biotype. We therefore treat the tree as being rooted on the long branch, which enables us to follow the sequence of events.
Among the El Tor isolates, the Australian clone is the most closely related of the current clones to the seventh pandemic clone, with 1 and 2 events along the 2 branches separating them. The Australian clone, although not discovered until 1977, must have arisen before the seventh pandemic and spread to Australia. The prepandemic outbreak isolates are located separately among the surviving El Tor pathogenic clones, with the 1937 Indonesian 66-2 (Makassar 759) isolate located closest to the seventh pandemic clone. The US Gulf clone diverged before the Australian clone and the seventh pandemic clones diverged, with a single recombination event on the branch to the common ancestor of the Australian and the seventh pandemic clones.
The 2 sixth pandemic isolates are well separated from the other strains, differing from them at 10 loci, an average of 5 events per branch. These include recombination events affecting 8 genes. If representative, ≈30% of the genes have undergone recombination during divergence of the sixth pandemic clone and the El Tor group of pathogenic clones that includes the seventh pandemic clone. The extensive divergence between the sixth pandemic and other toxigenic isolates studied indicates a long period since divergence from the common ancestor, which presumably occurred well before the sixth pandemic (1899–1923). In the absence of any intermediates, we cannot allocate individual events to either branch but presume that each is equally likely to have occurred on either branch.
This study using sequences of 26 genes has resolved the evolutionary relationship of the 2 major pandemic clones of V. cholerae and the relationships of the seventh pandemic clone to other pathogenic El Tor clones and isolates. With the relationships established it is clear that study of the prepandemic isolates and Australian clone in particular could illuminate the events involved in the emergence of the current seventh pandemic clone from this lineage.
Ms Salim is a PhD student in microbiology at the University of Sydney. Her research interests include population genetics and evolution of V. cholerae.
Acknowledgments
We thank all donors of isolates and the anonymous referees for their helpful suggestions.
This research was supported by grants from the National Health and Medical Research Council of Australia and a University of New South Wales Goldstar award.
References
- Pollitzer R. Cholera. Geneva: World Health Organization; 1959. p. 147–58.
- Lacey SW. Cholera: calamitous past, ominous future. Clin Infect Dis. 1995;20:1409–19. DOIPubMedGoogle Scholar
- Shimada T, Arakawa E, Itoh K, Okitsu T, Matsushima A, Asai Y, Extended serotyping scheme for Vibrio cholerae. Curr Microbiol. 1994;28:175–8. DOIGoogle Scholar
- Kotetishvili M, Stine OC, Chen Y, Kreger A, Sulakvelidze A, Sozhamannan S, Multilocus sequence typing has better discriminatory ability for typing Vibrio cholerae than does pulsed-field gel electrophoresis and provides a measure of phylogenetic relatedness. J Clin Microbiol. 2003;41:2191–6. DOIPubMedGoogle Scholar
- Barua D. History of cholera. In: Barua D, Greenough III WB, editors. Cholera. New York: Plenum; 1992. p. 1–36.
- Safa A, Bhuiyan NA, Alam M, Sack DA, Nair GB. Genomic relatedness of the new Matlab variants of Vibrio cholerae O1 to the classical and El Tor biotypes as determined by pulsed-field gel electrophoresis. J Clin Microbiol. 2005;43:1401–4. DOIPubMedGoogle Scholar
- Desmarchelier PM, Wong FY, Mallard K. An epidemiological study of Vibrio cholerae O1 in the Australian environment based on rRNA gene polymorphisms. Epidemiol Infect. 1995;115:435–46. DOIPubMedGoogle Scholar
- Johnston JM, Martin DL, Perdue J, McFarland LM, Caraway CT, Lippy EC, Cholera on a Gulf Coast oil rig. N Engl J Med. 1983;309:523–6. DOIPubMedGoogle Scholar
- Bhattacharya MK, Bhattacharya SK, Garg S, Saha PK, Dutta D, Nair GB, Outbreak of Vibrio cholerae non-O1 in India and Bangladesh. Lancet. 1993;341:1346–7. DOIPubMedGoogle Scholar
- Wachsmuth IK, Olsvik Ø, Evins GM, Popovic T. Molecular epidemiology of cholera. In: Wachsmuth IK, Blake PA, Olsvik Ø, editors. Vibrio cholerae and cholera, molecular to global perspectives. Washington: ASM Press; 1994. p. 357–70.
- Byun R, Elbourne LDH, Lan R, Reeves PR. Evolutionary relationships of the pathogenic clones of Vibrio cholerae by sequence analysis of four housekeeping genes. Infect Immun. 1999;67:1116–24.PubMedGoogle Scholar
- Farfan M, Minana-Galbis D, Fuste MC, Loren JG. Allelic diversity and population structure in Vibrio cholerae O139 Bengal based on nucleotide sequence analysis. J Bacteriol. 2002;184:1304–13. DOIPubMedGoogle Scholar
- Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature. 2000;406:477–83. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 11, Number 11—November 2005
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Peter R. Reeves, School of Molecular and Microbial Biosciences, G08, University of Sydney, Sydney, New South Wales 2006, Australia; fax: 61-2-9351-4571
Top