Volume 11, Number 12—December 2005
Research
Pandemic Strain of Foot-and-Mouth Disease Virus Serotype O
Abstract
A particular genetic lineage of foot-and-mouth disease virus (FMDV) serotype O, which we have named the PanAsia strain, was responsible for an explosive pandemic in Asia and extended to parts of Africa and Europe from 1998 to 2001. In 2000 and 2001, this virus strain caused outbreaks in the Republic of Korea, Japan, Russia, Mongolia, South Africa, the United Kingdom, Republic of Ireland, France, and the Netherlands, countries which last experienced FMD outbreaks decades before (ranging from 1934 for Korea to 1984 for the Netherlands). Although the virus has been controlled in all of these normally FMD-free or sporadically infected countries, it appears to be established throughout much of southern Asia, with geographically separated lineages evolving independently. A pandemic such as this is a rare phenomenon but demonstrates the ability of newly emerging FMDV strains to spread rapidly throughout a wide region and invade countries previously free from the disease.
Foot-and-mouth disease virus (FMDV, family Picornaviridae, genus Aphthovirus) causes an acute vesicular disease of pigs and wild and domesticated ruminants such as cattle, water buffalo, sheep, goats, and deer (1). It can cause high death rates in young animals and production losses in adults and is considered to be the single most important constraint to world trade in live animals and animal products. Spread of FMDV is predominantly associated with the legal and illegal movement of infected animals or their products.
The Food and Agriculture Organization World Reference Laboratory for Foot-and-Mouth Disease (WRLFMD) is established within the high-security laboratory at the Institute for Animal Health, Pirbright, United Kingdom (2). From 2000 to 2004, WRLFMD received an annual average of 536 samples to diagnose FMD from regions of the world where the disease is endemic, predominantly Africa and Asia. Seven serotypes of FMDV exist: SAT 1, SAT 2, and SAT 3 are usually restricted to Africa; Asia 1 is restricted to Asia; and O, A, and C are present in Africa, Asia, and South America and occasionally Europe. In each of the last 5 years, serotype O has been isolated from >60% of the positive FMD samples received.
The economic consequences of FMD incursion into disease-free regions may be severe. For instance, in the first 3 months of the 1997 outbreak in Taiwan, >6,000 farms were affected, 4 million pigs were destroyed or died from the disease, and >21 million doses of vaccine were used (3). The cost of controlling the disease was estimated at US $378.6 million. An additional $1.6 billion was lost in export trade, and >65,000 jobs in pig farming and associated industries were lost (3). To control the FMD outbreak without using vaccination, animals were slaughtered on >10,000 farms in the United Kingdom in 2001; only one fifth of these animals were actually infected. Four million animals were slaughtered for control measures and 2.5 million more for animal health reasons (4). The direct and indirect losses were estimated at ≈£8 billion (5).
FMDV has a genome consisting of a single strand of positive-sense RNA. Consequently, the virus has a high mutation rate and may change, on a random basis, 1–8 nucleotides (nt) per replication cycle (6). Nucleotide sequencing of part or all of the genome region coding for the outer capsid polypeptide VP1 was first used to study the epidemiology of FMD by Beck and Strohmaier (7), who investigated the origin of outbreaks of types O and A in Europe over a 20-year period. Since then, genetic variability has been used to individually characterize strains of FMDV and track their movement across international borders (8), and a large number of epidemiologic studies have been published (9). Previously, on the basis of comparisons of partial VP1 sequences (≈170 nt at the 3´ end of the gene) of FMD type O viruses, differences between 2 isolates within 4% have been suggested to indicate a recent common origin, whereas differences of >15% signify geographic isolation over many years (10), similar to the distinctions made between human polioviruses (11). Isolates with >85% nt sequence identity have been placed within groups or topotypes, which tend to be restricted in their geographic distribution (10,12). The 10 topotypes have been named Europe-South America (Euro-SA), Middle East–South Asia (ME-SA), Southeast Asia (SEA), Cathay (CHY), West Africa (WA), East Africa 1 (EA-1), East Africa 2 (EA-2), East Africa 3 (EA-3), Indonesia-1 (ISA-1), and Indonesia-2 (ISA-2). The Indonesian topotypes, which have not been identified since 1983, are considered extinct.
Knowles et al. (13) described the emergence and spread of the PanAsia strain from 1990 to 2000 on the basis of comparisons of partial (and some complete) VP1 sequences from 60 virus isolates. This article extends the molecular epidemiology of this virus strain by comparing 188 complete VP1 sequences for FMD type O viruses mostly isolated from 2000 to 2005 with published sequences of selected viruses from the previous decade and some reference virus strains (N = 151).
Viruses and Primers
The designation and origin of FMDV isolates studied are listed in Table A1.
Three alternative primer combinations were used for reverse transcription–polymerase chain reaction (RT-PCR): O-1C244F/NK61, O-1C272F/NK61, and O-1C283F/NK61, which have amplicon sizes of 1,181, 1,153, and 1,142 bp, respectively (Table). Forward and reverse primer amounts were 20 and 40 pmol, respectively. We used 4–6 internal sequencing primers to ensure coverage of the VP1 region on both DNA strands (Table).
RT-PCR of vRNA
Total RNA was extracted from 460 μL of a 10% epithelial suspension or cell culture supernatant by using RNeasy kits (Qiagen Ltd., Crawley, West Sussex, UK), according to the manufacturer's instructions, and resuspended in 50 μL nuclease-free water. This RNA (5 μL) was used as the template in a 1-step RT-PCR (Ready-To-Go RT-PCR Beads; Amersham Pharmacia Biosciences, Chalfont St. Giles, Bucks, UK). The following thermal profile was used: 42°C for 30 min; 94°C for 5 min; 35 cycles of 94°C for 60 s; 60°C for 60 s; and 72°C for 90 s; followed by a final extension of 72°C for 5 min. PCR products were analyzed by electrophoresis on a 1.5% agarose-Tris-borate-EDTA gel containing 0.5 μg/mL ethidium bromide. DNA weight markers (GeneRuler 100 bp DNA Ladder Plus, Ready-To-Use; Fermentas, Inc., Hanover, MD, USA) were run alongside the samples to facilitate product identification and quantification. Post-PCR removal of deoxynucleoside triphosphates and primers was achieved enzymatically by using ExoSAP-IT (USB Corporation, Cleveland, OH, USA), according to the manufacturer's instructions.
Sequence Determination
PCR amplicons were sequenced by using the DTS Quick Start Kit (Beckman Coulter Inc., Fullerton, CA, USA) according to the manufacturer's instructions and with the sequencing primers listed in the Table. The sequencing reactions were run on a CEQ8000Automated Sequencer (Beckman Coulter) according to the manufacturer's instructions. The sequences determined in this study have been submitted to the EMBL/GenBank/DDBJ databases; accession numbers are shown in Table A1.
Phylogenetic Analysis
An unrooted neighbor-joining tree was constructed by using MEGA version 3 (14). The robustness of the tree topology was assessed with 1,000 bootstrap replicates as implemented in the program.
Virus RNA was extracted from 188 FMD type O viruses, and each VP1-coding region was successfully amplified by RT-PCR by using at least 1 of the 3 described primer sets. The complete VP1 sequences were determined by directly sequencing the amplicons. For all these isolates, the VP1 gene consisted of 633 nt coding for 211 amino acids (previously VP1 was considered to be 2 amino acids longer at its carboxyl-terminus; however, the VP1-2A cleavage site is actually between a conserved glutamine [VP1211 in most type Os] and a variable residue [2A1, often a leucine in serotype O]) (15).
Figure 1

Figure 1. Midpoint–rooted neighbor-joining tree showing the relationships between the 339 VP1 sequences studied. Only the tree structure is shown; details of the boxes labeled A to C are shown in
Figure 2
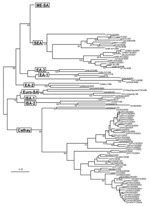
Figure 2. Midpoint-rooted neighbor-joining tree showing the Cathay, Europe-South America (Euro-SA), Indonesia-1 (ISA-1), Indonesia-2 (ISA-2), West Africa (WA), East Africa 1 (EA-1), East Africa 2 (EA-2), and East Africa 3 (EA-3) topotypes. Only...
Figure 4
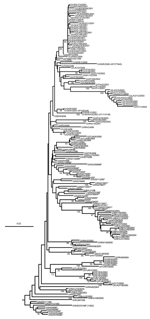
Figure 4. Midpoint-rooted neighbor-joining tree showing the PanAsia strain. Only bootstrap values >70% are shown.
The 188 VP1 sequences we report were compared to 151 VP1 sequences previously published or awaiting publication (database accession numbers are listed in Table A1). A bootstrapped neighbor-joining tree containing all 339 sequences was constructed by using MEGA 3 (Figure 1). Figures 2–4 show various parts of the tree depicted in Figure 1 in greater detail. The bootstrap support for the 10 FMDV O topotypes was generally high (96%–100%; Figure 2). The topotype distributions of the 299Asian FMD type O viruses (including those reported elsewhere) were as follows: ME-SA (253), SEA (18), and Cathay (49) (Table A1). Additionally, 26 European viruses (from the United Kingdom, Ireland, and France) belonged to the ME-SA topotype. The PanAsia strain accounted for 168 (66%) of the 253 ME-SA isolates.
Some FMDV O topotypes had a more limited spread than the ME-SA topotype. Virus isolates from Hong Kong and the Philippines all fell within the Cathay topotype; all the recently isolated (2000–2004) Philippines isolates form a distinct lineage. This topotype was first introduced into the Philippines in 1994, probably from mainland China or Hong Kong (the only known places where it existed at that time). Earlier isolates from the Philippines (e.g., O/PHI/5/95) were closely related to Hong Kong viruses (Figure 2). This topotype was first seen in Vietnam in 1997 and continued to occur there until 2004 (Figure 2) but has not, as far as we know, spread to neighboring Southeast Asian countries. A Cathay topotype virus also spread to Taiwan in 1997, where it caused an extensive epidemic that lasted until at least 1999 (3) (Figure 2). Viruses belonging to the SEA topotype continue to be isolated throughout Southeast Asia (Figure 2; Table A2), despite the recent introduction and widespread dissemination of the PanAsia strain. No examples of either of the Indonesian topotypes have been detected in the field since 1983.
Figure 3
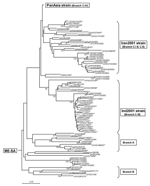
Figure 3. Midpoint-rooted neighbor-joining tree showing the Middle East–South Asia (ME-SA) topotype (except the PanAsia strain). Only bootstrap values >70% are shown.
Viruses belonging to the ME-SA topotype occur in many genetic sublineages (Figure 3). These were often initially found in India and subsequently spread to other geographic regions. The reference/vaccine strains (O5/IND/1/62, O1/Manisa/TUR/69, O1/Sharquia/EGY/72, and O/IND/R2/75) all occur in a single lineage distinct from later isolates. The O5/IND/1/62 sequenced by Hemadri et al. (16) is different (9.6%) from the same strain that we and others sequenced (17,18) (all 3 sequences are identical, and the virus stocks probably all originated from WRLFMD), and the origin of these isolates requires further investigation. Two other reference/vaccine strains (O/Geshur/ISR/85 and O/Dalton/ISR/2/88) fall on another lineage but are not closely related to each other. Within the ME-SA topotype, several sublineages have been defined as strains, such as PanAsia, Ind2001, and Iran2001, on the basis of phylogenetic relationships and a nucleotide difference of <5% (9,16). However, these are artificial groupings, the edges of which become blurred as viruses evolve in different directions. For example, the nucleotide sequences of 2 viruses that are on the PanAsia lineage, O/VIT/1/2004 and O/BHU/27/2004, differ from O/TAW/2/99 by 5.4% and 5.0%, respectively, but differ from each other by 7.9%. Thus trying to define "strains," particularly using percentage nucleotide relationships, may not be relevant, except in special circumstances, such as a pandemic caused by a cluster of closely related viruses.
Viruses that we consider part of the PanAsia strain (within the ME-SA topotype) are shown in Figure 4. Within the PanAsia strain, different sublineages can be distinguished despite some low bootstrap values. Some of them correspond to well-defined geographic areas in which these isolates have been collected through the years and show evolutionary relationships. Others are mixtures of FMDV isolates from different regions. In such cases, the phylogeny gives clues to the probable source of some isolates. The PanAsia strain shows a limited degree of variability of the VP1 gene during the outbreak in 2001 in the United Kingdom. Indeed, the degree of genetic variability of the VP1 gene of 24 isolates collected between the beginning and the end of the outbreak was <1.29%, and very few amino acid changes were observed (a maximum of 3 in any 1 sequence).
According to our current analysis, the PanAsia strain is an emergent sublineage of FMDV that, after several years in India, spread through southern Asia, the Middle East, and Europe. This strain apparently was confined to India for longer—and then spread much faster—than previously believed. In 1994, Samuel et al. (19) first noted the arrival of a new FMDV type O lineage in Saudi Arabia. Previously, we had considered this lineage to be part of the PanAsia strain (13). However, analysis of complete VP1 sequences with the neighbor-joining algorithm, rather than unweighted pair-group method analysis on partial VP1 sequences, indicated that these viruses, along with others isolated between 1994 and 1997 in Asia (except India), actually belong to 1 of 2 distinct lineages that we have termed Ind2001 and Iran2001 (Figure 3). Therefore, viruses that we would now classify as PanAsia first appeared in Bahrain, Iran, Lebanon, Kuwait, Saudi Arabia, and Yemen much later (i.e., in 1998); in Israel, Turkey, and the United Arab Emirates in 1999; and in Malaysia in 2000 (Figures 3 and 4; data not shown). In Nepal in 1990, viruses were found that were closely related to the earliest PanAsia isolates from India in the same year. However, from 1991 to 1996, only viruses belonging to non-PanAsia lineages of ME-SA were found in Nepal. During the years 1997–1999, PanAsia viruses were once again found. This virus lineage may have persisted in Nepal in the intervening years (since only a few virus isolates have been examined) or may have been reintroduced in 1997. This extension and reanalysis of the sequence data indicate that the spread of the PanAsia strain from the Indian subcontinent was probably more explosive than once thought and principally occurred from 1998 to 2001.
Retrospective examination of viruses from India indicated that the PanAsia strain was present in the north of that country as early as 1990 and may even have been present as far back as 1982 (16). From 1991 to 1997, the new lineage appeared to spread to other parts of India (16).
The presumed initial spread from India in 1998 was to Bhutan, Bahrain, Iran, Jordan, Kuwait, Lebanon, Syria, Saudi Arabia, and the Yemen Arab Republic. In May 1999, the People's Republic of China reported FMD outbreaks in Tibet, Hainan, and Fujian Provinces (20). Sequencing viruses from the outbreaks in Tibet (O/CHA/1/99, O/CHA/2/99, and O/CHA/3/99) and Hainan (O/CHA/4/99) showed that they belonged to the new lineage (13) (Figure 4). In June 1999, FMDV was isolated from subclinically infected or carrier cattle in Kinmen Prefecture of Taiwan Province of China (POC) during routine surveillance. Sequence analysis of this isolate (O/TAW/2/99) showed it also belonged to the new lineage (Figure 4). Later that month, FMDV was detected in Tainan Prefecture on the main island of Taiwan, again in cattle showing no signs of disease. In January 2000, the first clinical cases in cattle were found in Taiwan (Yunlin and Chiayii Prefectures) and in February 2000, ≈71 young goats in Kaoshiung and Changhwa Prefectures died suddenly from FMD, although no disease was seen in adult goats that had been vaccinated. The distribution of this sublineage throughout Asia justified its name of the PanAsia strain.
Towards the end of 1999, the PanAsia virus was clearly moving into Southeast Asia (Myanmar, Thailand, Vietnam, Lao People's Democratic Republic) (Table A2), where the FMDV type O SEA topotype had existed exclusively (at least until the Cathay topotype was introduced into Vietnam in 1997) (10). By April 2000, all mainland Southeast Asian countries had experienced outbreaks due to the new strain.
In March 2000, FMD type O appeared in South Korea and Japan, and sequence analysis indicated that the PanAsia strain was responsible (13) (Figure 4). In April 2000, a severe outbreak of FMD type O in occurred in pigs in the Ussuriysk District of eastern Russia. Of 625 pigs affected, nearly 37% died from the disease. Sequencing the VP1 gene showed that the PanAsia strain was responsible (13). At the end of April 2000, an outbreak of FMD type O was reported in Ulaanbadrakh Soum County, Dornogovi Province, Mongolia. In this outbreak sheep, goats, and cattle were affected. Again, sequence analysis of the VP1 gene showed the virus to be of the PanAsia lineage (13). In September 2000, the PanAsia strain spread to KwaZulu-Natal Province in South Africa (13,17) (Figure 4); the origin was traced to feeding pigs with uncooked swill from a ship in the port of Durban (21). This FMD outbreak is the first since 1957 in this region of South Africa and the first recorded outbreak in that country due to serotype O. In February 2001, FMD was diagnosed in the United Kingdom; by the end of July, >1,900 farms were affected. The PanAsia strain was responsible for these outbreaks (13,22,23). In late February 2001, the disease spread from the British mainland to Northern Ireland, and in March and April outbreaks of FMD type O were also reported in the Republic of Ireland (n = 1), France (n = 2), and the Netherlands (n = 26). In 2003, the PanAsia strain was detected for the first time in Afghanistan, Nepal, and Pakistan; however, because of lack of samples or sequencing data, the strain may have been present earlier. Since 2003, the PanAsia strain has not been detected in any new countries.
The PanAsia strain has not yet been detected in Africa (except South Africa in 2000) or South America, despite extensive unpublished sequence studies by ourselves; the Onderstepoort Veterinary Institute, South Africa (W. Vosloo, pers. comm.); and the Pan-American FMD Center, Brazil (I.E. Bergmann, pers. comm.). However, the PanAsia strain is present in many countries in which FMD is endemic and occurs in countries in which the incidence of FMD is sporadic.
The extent of this spread is unique for a single strain of FMDV, and its presence in most recent samples from the Middle East indicates that it has dominated and outcompeted the other strains of FMDV previously observed (19). While we acknowledge that the sampling of virus isolates is not random (i.e., the samples examined are those submitted to WRLFMD by some of the countries experiencing outbreaks), the same sampling technique has shown a marked increase in the number of isolations of the PanAsia lineage over the preceding years.
The appearance of the PanAsia virus in countries that have been FMD-free for many years shows that this strain is capable of spreading to countries where strict control measures are normally effective at preventing importation of animal pathogens. Whether this fitness to survive is related to particular features of the transmissibility of the virus strain or its ability to spread subclinically in certain breeds of animal, as found in Taiwan in 1999 or in Japan in 2000 (24), is not clear. The PanAsia virus strain has been isolated from a wide variety of host species, including cattle, water buffalo, pigs, sheep, goats, and gazelle (Qatar in 1999), and its ability to infect a wide range of species could be a contributing factor in its success. Within the PanAsia strain, differences in behavior of the virus, such as host species or virulence, remain unexplained on a genetic basis, according to comparison of the full genome sequences from viruses from this group (25). However, these characteristics can also be biased by practices such as vaccination, the animal population targeted for vaccination, or the animal species that are farmed in a particular area.
We have no evidence of increased or altered trade in the region that could explain the sudden spread of the PanAsia virus. Additionally, the lack of efficacy of existing FMDV vaccines does not seem to be responsible for the spread of this strain in countries in which vaccination is practiced. Indeed, antigenic matching analysis has shown good cross-reactivity between field isolates of the PanAsia strain and current vaccine strains such as O1 Manisa (WRLFMD, data not shown), and this finding has been confirmed for O/UKG/2001 virus by cross-protection studies (26,27).
The spread of the PanAsia strain across most of Asia and into Europe and South Africa demonstrates how a newly evolved virus may become established, in spite of control measures at international borders. FMD in a previously disease-free country can seriously interfere with the local and export trade in susceptible animals and their products. A large outbreak of FMD in northern Europe or the United States could result in losses of several billion US dollars. The emergence of this strain of FMDV, and its spread within the territory bounded by Ireland in the west and Japan in the east, provides an example of the economic damage that can result. It also demonstrates the difficulty of containing such a transmissible virus within a defined region. The emergence of such strains highlights the necessity to constantly monitor and characterize field isolates responsible for outbreaks in FMD-endemic countries and the need for countries to be rapidly alerted so that appropriate control measures can be instituted. For this purpose, an international early warning system must be established to share information on the characteristics of the latest FMDV isolates in real time.
Mr Knowles is a molecular virologist at the Institute for Animal Health. His research interests focus on the molecular epidemiology and evolution of picornaviruses of animals, particularly FMDV.
Acknowledgments
We acknowledge the work of Nigel Ferris and colleagues in receiving and serotyping the viruses submitted to WRLFMD.
This work was supported by the Department for Environment, Food and Rural Affairs, United Kingdom (Reference Laboratory contract and research grant no. SE 2921). The submission and serotyping of samples were supported by DEFRA and a grant from the European Commission for the Control of FMD.
References
- Thomson GR. Foot-and-mouth disease. In: Coetzer JAW, Thomson G R, Tustin RC, Kriek NPJ, editors. Infectious diseases of livestock with special reference to southern Africa. Vol. 2. Cape Town: Oxford University Press; 1994. p. 825–52.
- Ferris NP, Donaldson AI. The World Reference Laboratory for Foot-and-Mouth Disease: a review of thirty-three years of activity (1958-1991). Rev Sci Tech. 1992;11:657–84.PubMedGoogle Scholar
- Yang PC, Chu RM, Chung WB, Sung HT. Epidemiological characteristics and financial costs of the 1997 foot-and-mouth disease epidemic in Taiwan. Vet Rec. 1999;145:731–4.PubMedGoogle Scholar
- Scudamore JM, Harris DM. Control of foot-and-mouth disease: lessons from the experience of the outbreak in Great Britain in 2001. Rev Sci Tech. 2002;21:699–710.PubMedGoogle Scholar
- Thompson D, Muriel P, Russell D, Osborne P, Bromley A, Rowland M, Economic costs of the foot-and-mouth disease outbreak in the United Kingdom in 2001. Rev Sci Tech. 2002;21:675–87.PubMedGoogle Scholar
- Domingo E, Dopazo J, Rodrigo MJ, Rodriguez A, Saiz JC, Sabrino F. Aphthovirus evolution. In: Gibbs AJ, Calisher CH, Garcia-Arenal F, editors. Molecular basis of virus evolution. Cambridge (UK): Cambridge University Press; 1995. p. 310–20.
- Beck E, Strohmaier K. Subtyping of European foot-and-mouth disease virus strains by nucleotide sequence determination. J Virol. 1987;61:1621–9.PubMedGoogle Scholar
- Knowles NJ, Samuel AR. Molecular techniques in foot-and-mouth disease epidemiology. Towards livestock disease diagnosis and control in the 21st century: proceedings of an international symposium on diagnosis and control of livestock diseases jointly organized by the International Atomic Energy Agency and the Food and Agriculture Organization of the United Nations. Vienna, 1997 Apr 7–11. Vienna: International Atomic Energy Agency; 1998. p. 185–201.
- Knowles NJ, Samuel AR. Molecular epidemiology of foot-and-mouth disease virus. Virus Res. 2003;91:65–80. DOIPubMedGoogle Scholar
- Samuel AR, Knowles NJ. Foot-and-mouth disease type O viruses exhibit genetically and geographically distinct evolutionary lineages (topotypes). J Gen Virol. 2001;82:609–21.PubMedGoogle Scholar
- Rico-Hesse R, Pallansch MA, Nottay BK, Kew OM. Geographic distribution of wild poliovirus type 1 genotypes. Virology. 1987;160:311–22. DOIPubMedGoogle Scholar
- Knowles NJ, Davies PR, Midgley RJ, Valarcher J-F. Identification of a ninth foot-and-mouth disease virus type O topotype and evidence for a recombination event in its evolution. Report of the Session of the Research Group of the Standing Technical Committee of EUFMD, Chania, Crete, Greece, 2004 Oct 12–15. Rome: Food and Agriculture Organization; 2004. Appendix 24, p. 163–72.
- Knowles NJ, Samuel AR, Davies PR, Kitching RP, Venkataramanan R, Kanno T, Emergence of a pandemic strain of foot-and-mouth disease virus serotype O. Report of the Session of the Research Group of the Standing Technical Committee of the European Commission for the Control of Foot-and-Mouth Disease, Borovets, Bulgaria, 2000 Sep 5–8. Rome: Food and Agriculture Organization; 2000. Appendix 1. p. 20–31.
- Kumar S, Tamura K, Nei M. MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform. 2004;5:150–63. DOIPubMedGoogle Scholar
- Birtley JR, Knox SR, Jaulent AM, Brick P, Leatherbarrow RJ, Curry S. Crystal structure of foot-and-mouth disease virus 3C protease. New insights into catalytic mechanism and cleavage specificity. J Biol Chem. 2005;280:11520–7. DOIPubMedGoogle Scholar
- Hemadri D, Tosh C, Sanyal A, Venkataramanan R. Emergence of a new strain of type O foot-and-mouth disease virus: its phylogenetic and evolutionary relationship with the PanAsia pandemic strain. Virus Genes. 2002;25:23–34. DOIPubMedGoogle Scholar
- Sangare O, Bastos AD, Marquardt O, Venter EH, Vosloo W, Thomson GR. Molecular epidemiology of serotype O foot-and-mouth disease virus with emphasis on West and South Africa. Virus Genes. 2001;22:345–51. DOIPubMedGoogle Scholar
- Carrillo C, Tulman ER, Delhon G, Lu Z, Carreno A, Vagnozzi A, Comparative genomics of foot-and-mouth disease virus. J Virol. 2005;79:6487–504. DOIPubMedGoogle Scholar
- Samuel AR, Knowles NJ, Kitching RP, Hafez SM. Molecular analysis of foot-and-mouth disease type O viruses isolated in Saudi Arabia between 1983 and 1995. Epidemiol Infect. 1997;119:381–9. DOIPubMedGoogle Scholar
- Foot and mouth disease in the People's Republic of China. OIE Disease Information. 1999;12:76.
- Foot and mouth disease in South Africa. OIE Disease Information. 2000;13:164–5.
- Knowles NJ, Samuel AR, Davies PR, Kitching RP, Donaldson AI. Outbreak of foot–and–mouth disease virus serotype O in the UK caused by a pandemic strain. Vet Rec. 2001;148:258–9.PubMedGoogle Scholar
- Samuel AR, Knowles NJ. Foot-and-mouth disease virus: cause of the recent crisis for the UK livestock industry. Trends Genet. 2001;17:421–4. DOIPubMedGoogle Scholar
- Sakamoto K, Yamakawa M, Kanno T, Murakami Y. Pathogenesis of O/JPN/2000 to susceptible animals. Report of the Session of the Research Group of the Standing Technical Committee of the European Commission for the Control of Foot-and-Mouth Disease, Borovets, Bulgaria, 2000 Sept 5–8. Rome: Food and Agriculture Organization; 2000. Appendix 2. p. 32–8.
- Mason PW, Pacheco JM, Zhao Q-Z, Knowles NJ. Comparisons of the complete genomes of Asian, African and European isolates of a recent foot-and-mouth disease virus type O pandemic strain (PanAsia). J Gen Virol. 2003;84:1583–93. DOIPubMedGoogle Scholar
- Aggarwal N, Zhang Z, Cox S, Statham R, Alexandersen S, Kitching RP, Experimental studies with foot-and-mouth disease virus, strain O, responsible for the 2001 epidemic in the United Kingdom. Vaccine. 2002;20:2508–15. DOIPubMedGoogle Scholar
- Cox SJ, Voyce C, Parida S, Reid SM, Hamblin PA, Paton DJ, Protection against direct-contact challenge following emergency FMD vaccination of cattle and the effect on virus excretion from the oropharynx. Vaccine. 2005;23:1106–13. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 11, Number 12—December 2005
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Nick J. Knowles, Institute for Animal Health, Pirbright Laboratory, Ash Rd, Pirbright, Woking, Surrey, GU24 0NF, UK; fax: 44-1483-232448
Top