Volume 11, Number 2—February 2005
Research
Novel Flavivirus or New Lineage of West Nile Virus, Central Europe
Abstract
A flavivirus (strain 97-103) was isolated from Culex pipens mosquitoes in 1997 following floods in South Moravia, Czech Republic. The strain exhibited close antigenic relationship to West Nile virus (WNV) prototype strain Eg-101 in a cross-neutralization test. In this study, mouse pathogenicity characteristics and the complete nucleotide and putative amino acid sequences of isolate 97-103, named Rabensburg virus (RabV) after a nearby Austrian city, were determined. RabV shares only 75%–77% nucleotide identity and 89%–90% amino acid identity with representative strains of WNV lineages 1 and 2. Another RabV strain (99-222) was isolated in the same location 2 years later; it showed >99% nucleotide identity to strain 97-103. Phylogenetic analyses of RabV, WNV strains, and other members of the Japanese encephalitis virus (JEV) complex clearly demonstrated that RabV is either a new (third) lineage of WNV or a novel flavivirus of the JEV group.
West Nile virus (WNV), a member of the Japanese encephalitis virus (JEV) group within the genus Flavivirus, family Flaviviridae, is the most widespread flavivirus, occurring in Africa, Eurasia, Australia, and North America. Other members of the JEV group flaviviruses are Cacipacore virus (CPCV), Koutango virus (KOUV), JEV, Murray Valley encephalitis virus (MVEV), Alfuy virus (ALFV), St. Louis encephalitis virus (SLEV), Usutu virus (USUV), and Yaounde virus (YAOV) (1). Although initially WNV was considered to have minor human health impact, the human and equine outbreaks in Europe (Romania, Russia, France, Italy), Africa (Algeria, Tunisia, Morocco), and Asia (Israel) within the last 10 years, and especially the virus’s emergence and spread in North America since 1999, put it into the focus of scientific interest. The distribution and ecology of WNV, as well as clinical features, pathogenesis, and epidemiology of West Nile disease have been reviewed (2–6). Phylogenetic analyses showed 2 distinct lineages of WNV strains, (which themselves subdivide into several subclades or clusters, isolated in different geographic regions (7–10).
The presence of WNV in central Europe has been known for a long time. Serologic surveys have detected specific antibodies to WNV in several vertebrate hosts in Austria, Czech Republic, Hungary, and Slovakia during the past 40 years, and several virus strains were isolated from mosquitoes, rodents, and migrating birds (3). Human cases of West Nile fever were reported in the Czech Republic in 1997 (11) and in Hungary in 2003 (12). Although these countries are important transit areas or final destinations for migratory birds from the African continent, and hence may play an important role in the circulation and conservation of different WNV strains, genetic information about the strains isolated in central Europe has not been available. We report the complete genome sequence and phylogenetic analyses, as well as antigenic and mouse virulence characteristics, of a unique flavivirus strain (97-103), closely related to WNV, which was isolated by intracranial injection of suckling mice with homogenates of female Culex pipiens mosquitoes collected 10 km from Lanzhot, Czech Republic, after a flood in 1997 (11,13,14). The collection site was very close to the Czech-Austrian border, ≈2 km from the small Austrian town of Rabensburg. Consequently, the isolate 97-103 was later tentatively called Rabensburg virus (RabV). Another antigenically identical or very closely related strain (99-222) was isolated from Cx. pipiens mosquitoes in the same location 2 years later (14).
Isolates 97-103 (passage 5 in suckling mouse brain [SMB]) and 99-222 (passage 4 in SMB) were freeze-dried in October 2000 (14). Viral RNA was extracted from 140 μL of virus resuspended in diethylpyrocarbonate (DEPC)-treated water, using the QIAamp viral RNA Mini Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions. For amplification of the complete genome, oligonucleotide primers were designed with the help of the Primer Designer 4 for Windows 95 program (Scientific and Educational Software, version 4.10) and were synthesized by GibcoBRL Life Technologies, Ltd. (Paisley, Scotland, UK). A complete list of the 35 primers used in reverse transcription–polymerase chain reaction (RT-PCR) and sequencing reactions is available upon request. Reverse transcription and amplification were performed with a continuous RT-PCR method with the Qiagen OneStep RT-PCR Kit (Qiagen) following the manufacturer’s instructions. Reverse transcription (at 50°C for 30 min) was followed by a denaturation step at 95°C for 15 min, and 40 cycles of amplification (94°C for 40 s, 57°C for 50 s, 72°C for 1 min). Reactions were completed by a final extension for 7 min at 72°C, and the amplicons were kept at 4°C until electrophoresis was carried out. The reactions were performed in a Perkin-Elmer GeneAmp PCR System 2400 thermocycler (Perkin-Elmer Corp., Wellesley, MA, USA). After RT-PCR, the amplicons were electrophoresed in agarose gel, stained with ethidium bromide, and bands were visualized under UV light. Gels were photographed with a Kodak DS Electrophoresis Documentation and Analysis System (Eastman Kodak Company, New Haven, CT, USA). Product sizes were determined with reference to a 100–bp DNA Ladder (Promega, Madison, WI, USA). Fluorescence-based direct sequencings were performed in both directions on the PCR products with the ABI Prism Big Dye Terminator cycle sequencing ready reaction kit (Perkin-Elmer) and an ABI Prism 310 genetic analyzer (Perkin-Elmer) automated sequencing system (15).
The nucleotide sequences were identified by BLAST search against GenBank databases and were compiled and aligned with the help of the Align Plus 4 for Windows 95 (Scientific and Educational Software, version 4.00) and ClustalX Multiple Sequence Alignment (version 1.81) programs. Phylogenetic analysis was performed with the Phylogeny Inference Program Package (PHYLIP) version 3.57c. Distance matrices were generated by the Fitch program, with a translation/transversion ratio of 2.0. Phylogenetic trees were delineated by using the TreeView (Win32) program version 1.6.6.
Both virus strains were identified as WNV by complement fixation and neutralization tests (11,13). Strain 97-103 was compared antigenically in detail with the Egyptian Eg-101 topotype strain of WNV (16), a representative of WNV lineage 1 (clade 1a). In plaque-reduction cross-neutralization tests (PRNT) with homologous and heterologous antisera (produced by injection of ICR mice with 3 intraperitoneal doses at weekly intervals), the serum raised against Eg-101 neutralized both the homologous virus and 97-103 at a titer of 512, while the strain 97-103 specific serum was effective against strain Eg-101 only at a titer of 64, although it neutralized the homologous virus at 512. The average 4-fold difference in cross-PRNT titers indicates certain antigenic heterogeneity of the 2 strains, and the 97-103 isolate was therefore regarded as “a subtype of WNV” (14).
Virulence of RabV strains 97-103 and 99-222 was determined by intracranial and intraperitoneal injection of specific-pathogen-free (SPF) outbred ICR mice. Central nervous system symptoms (e.g., pareses of hind limbs) developed in suckling mice, which died 7–15 days after intracranial injection (Table 1). Adult mice did not show any clinical symptoms and survived the experimental infection. On the other hand, the WNV topotype strain Eg-101 caused fatal illness in intracranially injected mice, killing them within 4 to 6 days after infection, regardless of their age (11,13). After intraperitoneal injection, strain Eg-101 killed all suckling mice but a <10% of adult mice; RabV strains 97-103 and 99-222 killed approximately one third of suckling mice, and the average survival time was 11 days (range 10–14 days). Thus, both Rabensburg virus strains exhibit clearly lower virulence for mice than the Egyptian WNV topotype strain. In addition, average survival time of suckling ICR mice injected intracranially with RABV was significantly longer than with strain Eg-101.
The genome of strain 97-103 Rabensburg virus (RabV) was investigated by RT-PCR and subsequent direct sequencing of the amplicons. Initially, oligonucleotide primers designed on the consensus sequences of linage 1 and 2 WNV strains were applied to the viral nucleic acid of RabV. On the basis of the sequence information obtained from these PCR products, specific primer pairs were designed to produce overlapping amplicons covering the entire genome. The RT-PCR products were sequenced, and the sequences were compiled, resulting in a 10,972 nucleotide (nt-) long sequence that represented the complete genome of the virus. The sequence was identified by BLAST search against GenBank databases. The highest identity rates of RabV to other flaviviruses (78%–90%) were found with certain regions of WNV strains of lineage 1 and 2.
From the second isolate (99-222), 5 genomic regions have been amplified and sequenced so far, showing a total of 3656 nt. They represent partial coding sections from the core (C), anchored C, premembrane (PreM), and membrane (M) protein regions (between nucleotide positions 117 and 752); NS3 protein region (between nucleotide positions 5294 and 5536, and between nucleotide positions 5565 and 6343); NS4b and NS5 regions (between nucleotide positions 7321 and 8112); and NS5 protein region (between nucleotide positions 9095 and 10305). Partial sequence analysis of isolate 99-222 showed >99% identity to 97-103. Aligned to strain 97-103, only a few nucleotide substitutions were observed, in the following positions: C609 to T; C720 to A; G5727 to A (resulting in an amino acid change Met to Ile); T5910 to C (resulting in an amino acid change Ile to Thr); T5961 to C; C9630 to A; and G9843 to T.
Similar to other flaviviruses (17), the nucleotide sequence of RabV contains l open reading frame (ORF) encoding the viral proteins as a large polyprotein precursor. The ORF starts at nucleotide position 97, and codes for a 3,433-amino acid (aa) polypeptide. The putative amino acid sequence of the polyprotein precursor gene of RabV 97-103 has been translated; based on the amino acid alignment with WNV, the putative mature proteins, conserved structural elements, and putative enzyme motifs were localized. The anchored C protein is located between nt 97 and 465; within this region, the C protein is located between nt 97 and 411. The PreM protein is encoded from nt 466 to nt 966, with the M protein between nt 742 and 966. The envelope (E) protein is encoded between nucleotide positions 967 and 2469, followed by the nonstructural proteins NS1 (nt 2470–3525), NS2a (nt 3526–4218), NS2b (nt 4219–4611), NS3 (nt 4612–6468), NS4a (nt 6469–6846), 2K (nt 6847–6915), NS4b (nt 6916–7680), and NS5 (nt 7681–10395), respectively. Amino acid identities with WNV were found at the known conserved positions (i.e., Cys residues involved in intramolecular bonds in the E and NS1 protein, putative integrin binding motif of the E protein, catalytic triad and substrate binding pocket of the trypsin-like serine protease, RNA helicase motif of the NS3 protein, and RNA-dependent RNA polymerase motif of the NS5 protein; 15).
To investigate the phylogenetic relationship of RabV to other WNV isolates, multiple nucleotide and putative amino acid sequence alignments were made involving 16 WNV strains (listed in Table 2). Although several complete WNV nucleotide sequences from previously published studies (10,18) have been deposited in the GenBank databases, only selected representatives of lineages and clades have been included in our alignments, in order to obtain more precise and demonstrative trees.
Figure 1
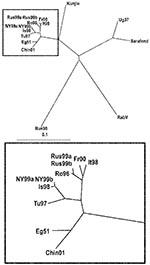
Figure 1. Phylogenetic tree illustrating the genetic relationship between selected West Nile virus strains based on their complete genome sequences. Bar on the left demonstrates the genetic distance. (Abbreviations and accession numbers are...
RabV exhibited 73%–77% nucleotide identity rates to the different WNV strains (Table 3). The relationships between the strains are demonstrated in Figure 1. The 2 lineages of WNV are obviously separated in the tree. Clade 1a viruses form a tight cluster with close genetic relationship among the members. Kunjin virus, the representative of clade 1b, appears as a separate branch of lineage 1. Unfortunately, no complete genome sequence information is available on clade 1c (Indian strains); thus, they are not represented in the tree. The prototype Uganda strain B956 (WNFCG) of lineage 2 is grouped together with the Sarafend strain, a laboratory strain with uncertain origin and passage history. Two viruses proved to be clearly distinct with significant genetic distances to all other WNV strains and also from each other: RabV and strain LEIV-Krnd88-190 (in the phylogenetic trees designated Rus98). The latter virus was isolated from Dermacentor marginatus ticks in the northwest Caucasus Mountain valley in 1998 and was regarded as a new variant of WNV (19–21). Because these 2 viruses differ considerably from all WNV strains, the issue is raised about whether classifying these 2 viruses as separate members of the JEV group might be more appropriate.
Figure 2
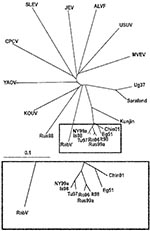
Figure 2. Phylogenetic tree illustrating the genetic relationship between representatives of the Japanese encephalitis virus complex and selected West Nile virus strains based on partial genome sequences of the NS5 protein gene. Bar...
To elucidate this question, a comprehensive phylogenetic analysis was performed on all representatives of the JEV group. Because only partial common sequence information of the NS5 protein gene region is currently available from SLEV, ALV, CPCV, KOUV, and YAOUV (22), the phylogenetic analysis had to be restricted to this region (Figure 2). Within the investigated genome stretch, RabV showed 77%–78% identity to lineage 1 and 2 WNV strains, 77% identity to strain LEIV-Krnd88-190, and 71%–76% identity to other representatives of the JEV group. In the phylogenetic tree (Figure 2), the separation of the 2 unique strains (RabV and LEIV-Krnd88-190 = Rus98) from WNV is clearly visible. Although RabV exhibits the closest relationship to the WNV representatives, similar identity rates (76%) exist between MVEV and USUV, as well as between JEV and ALFV, and these viruses have been taxonomically classified as separate viruses. The Rus98 virus clusters together with KOUV, a virus isolated originally from a Kemp’s gerbil (Tatera kempi) in Senegal 1968 and subsequently recovered from other rodent species and several genera of ticks (Rhipicephalus, Hyalomma, Alectorobius) in central Africa (23). The Rus98 strain was also isolated from ticks.
The putative amino acid sequence of RabV was also compared with the corresponding sequences of representatives of WNV lineages and clades, as well as with other JEV group viruses on the available polypeptide sequence regions. RabV shared 89%–90% identity on the complete polypeptide precursor region with the WNV strains, 87% identity with the Rus89 strain, and 75%–76% identity with JEV, USUV, and MVEV. The alignments of the partial amino acid sequences of the NS5 region (between aa 2991 and 3335) showed 94%–96% identity rates with the WNV strains, 95% with strain Rus98, and 78%–90% with the other members of the JEV group (Table 3). Phylogenetic trees, based on the amino acid alignments, displayed nearly identical topology to nucleotide sequence–based trees (data not shown). The complete genome sequence of RabV (flavivirus strain 97-103) has been deposited in GenBank under accession no. AY765264.
WNV strains of different lineages exhibit considerable genomic diversity (76%–77% nucleotide identity only). At the same time, WNV is not sharply delimited genomically from the other members of the JEV group. The available partial sequences of the NS5 gene region from other viruses of the group show 71%–76% nucleotide and 78%–90% amino acid identities to WNV strains. The closest relatives of WNV are KOUV and YAOV (10,22–24).
Lineage 1 of WNV comprises strains from several continents and is subdivided into at least 3 clades. In clade 1a, several subclades or clusters are formed by closely related strains, such as strains isolated 40–50 years ago in Europe and Africa; strains isolated 20–30 years ago in Africa; strains isolated within the last 10 years in Europe and Africa; and strains isolated within the last 5 years in the United States and Israel. Clade 1b consists of the Australian isolates (Kunjin), while clade 1c contains strains from India. Lineage 2 is composed of WNV strains that have been isolated, so far exclusively, in the sub-Saharan region of Africa and in Madagascar (18). The genetic distance between the 2 lineages is relatively great in contrast to that within some representatives of lineage 1 that were isolated in distant geographic locations and within considerable time intervals. While the viruses in clade 1a share 95.2%–99.9% nucleotide and 99.3%–100% amino acid identity to each other, and also 86.6%–87.8% nucleotide and 97.4%–97.7% amino acid identity to the clade 1b viruses, the overall identity rates between lineage 1 and 2 are only 75.7%–76.8% on nucleotide level and 93.2%–94.0% on amino acid level (18), identity rates that resemble those between RabV and either lineage 1 or lineage 2 WNV strains. Besides genomic differences, antigenic variability can be observed in cross-neutralization analyses and monoclonal antibody binding assays (8,18).
The results of the phylogenetic analyses indicate that viruses closely related to WNV are present in central Europe and southern Russia. Although these viruses have initially been identified as WNV, they can be regarded, on the basis of their genetic distances, either as separate lineages of WNV (RabV: lineage 3; LEIV-Krnd88-190 = Rus98: lineage 4) or as new viruses within the JEV group. The antigenic and biologic differences between RabV and the WNV reference strain Eg-101 also support this opinion. Isolation of RabV in 1997 was obviously not an isolated event; rather, flaviviruses of the RabV type seem to be present or persist in this area, as demonstrated by the isolation of an almost identical virus strain (99-222) 2 years later (14). The ecology of RabV needs further investigation. Other unanswered questions concern the pathogenicity and host spectrum of the virus, especially regarding possible human infections.
To summarize, a novel flavivirus strain of unknown human pathogenicity, repeatedly isolated from Cx. pipiens mosquitoes in central Europe, has been molecularly characterized, including determination of its complete nucleotide and deduced amino acid sequences. Based on the analysis of the virus and comparison with related viruses including phylogenetic relationships, we suggest that RabV be classified either as a new (third) lineage of WNV or as a novel flavivirus within the JEV group.
Dr. Bakonyi is a lecturer in virology at the Faculty of Veterinary Science, Budapest, and also works as a guest researcher at the University of Veterinary Medicine, Vienna. He is interested in the molecular diagnosis and epidemiology of animal and human viruses.
Acknowledgment
This study was funded by a grant of the Austrian Federal Ministry for Health and Women’s Issues, and it was also supported by the Czech Science Foundation (206/03/0726).
References
- Heinz FX, Collett MS, Purcell RH, Gould EA, Howard CR, Houghton M, Family Flaviviridae. In: van Regenmortel MHV, Faquet CM, Bishop DHL, editors. Virus taxonomy, Seventh International Committee for the Taxonomy of Viruses. San Diego: Academic Press; 2000. p. 859–78.
- Hayes CG. West Nile fever. In: Monath TP, editor. The arboviruses: epidemiology and ecology. Vol. V. Boca Raton (FL): CRC Press; 1989. p. 59–88.
- Hubálek Z, Halouzka J. West Nile fever—a reemerging mosquito-borne viral disease in Europe. Emerg Infect Dis. 1999;5:643–50. DOIPubMedGoogle Scholar
- Hubálek Z. European experience with the West Nile virus ecology and epidemiology: could it be relevant for the New World? Viral Immunol. 2000;13:415–26. DOIPubMedGoogle Scholar
- Murgue B, Zeller H, Deubel V. The ecology and epidemiology of West Nile virus in Africa, Europe. In: Mackenzie JS, Barrett ADT, Deubel V, editors. Japanese encephalitis and West Nile viruses. Current topics in Microbiology. Vol. 267: West Nile. Berlin: Springer; 2002. p. 195–221.
- Zeller HG, Schuffnecker I. West Nile virus: an overview of its spread in Europe and the Mediterranean basin in contrast to its spread in the Americas. Eur J Clin Microbiol Infect Dis. 2004;23:147–56. DOIPubMedGoogle Scholar
- Berthet F, Zeller HG, Drouet M, Rauzier J, Digoutte J, Deubel V. Extensive nucleotide changes and deletions within the envelope gene of Euro-African West Nile viruses. J Gen Virol. 1997;78:2293–7.PubMedGoogle Scholar
- Savage HM, Ceianu C, Nicolescu G, Karabatsos N, Lanciotti RS, Vladimirescu A, Entomologic and avian investigations of an epidemic of West Nile fever in Romania, 1996, with serologic and molecular characterization of a virus from mosquitoes. Am J Trop Med Hyg. 1999;61:600–11.PubMedGoogle Scholar
- Lanciotti RS, Roehrig JT, Deubel V, Smith J, Parker M, Steele K, Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern U.S. Science. 1999;286:2333–7. DOIPubMedGoogle Scholar
- Charrel RN, Brault AC, Gallian P, Lemasson JJ, Murgue B, Murri S, Evolutionary relationship between Old World West Nile virus strains evidence for viral gene flow between Africa, the Middle East, and Europe. Virology. 2003;315:381–8. DOIPubMedGoogle Scholar
- Hubálek Z, Halouzka J, Juricova Z. West Nile fever in Czechland. Emerg Infect Dis. 1999;5:594–5. DOIPubMedGoogle Scholar
- Ferenzi E, Bakonyi T, Tóth-Mittler E, Czeglédi A, Bán E. Emergence of an old-new virus: domestic West Nile virus infections with central nervous system symptoms, 2003–2004. In: Abstracts of the 2004 year Congress of the Hungarian Society for Microbiology; 2004. Oct. 7–9, Keszthely, Hungary. Abstract p. 36–7 [in Hungarian]. Budapest: Hungarian Soceity for Microbiology; 2004.
- Hubálek Z, Halouzka J, Juricova Z, Sebesta O. First isolation of mosquito-borne West Nile virus in the Czech Republic. Acta Virol. 1998;42:119–20.PubMedGoogle Scholar
- Hubálek Z, Savage HM, Halouzka J, Juricova Z, Sanogo YO, Lusk S. West Nile virus investigations in South Moravia, Czechland. Viral Immunol. 2000;13:427–33. DOIPubMedGoogle Scholar
- Bakonyi T, Gould EA, Kolodziejek J, Weissenböck H, Nowotny N. Complete genome analysis and molecular characterization of Usutu virus that emerged in Austria in 2001; comparing with the South African strain SAAR-1776 and other flaviviruses. Virology. 2004;328:301–10. DOIPubMedGoogle Scholar
- Melnick JL, Paul JR, Riordan JT, Barnett VH, Goldblum N, Zabin E. Isolation from human sera in Egypt of a virus apparently identical to West Nile virus. Proc Soc Exp Biol Med. 1951;77:661–5.PubMedGoogle Scholar
- Rice CM, Lenches EM, Eddy SR, Shin SJ, Sheets RL, Strauss JH. Nucleotide sequence of yellow fever virus: Implications for flavivirus gene expression and evolution. Science. 1985;229:726–33. DOIPubMedGoogle Scholar
- Lanciotti RS, Ebel GD, Deubel V, Kerst AJ, Murri S, Meyer R, Complete genome sequences and phylogenetic analysis of West Nile virus strains isolated from the United States, Europe, and the Middle East. Virology. 2002;298:96–105. DOIPubMedGoogle Scholar
- Prilipov AG, Kinney RM, Samokhvalov EI, Savage HM, Al’khovskii SV, Tsuchiya KR, Analysis of new variants of West Nile fever virus [in Russian]. Vopr Virusol. 2002;47:36–41.PubMedGoogle Scholar
- Lvov DK, Kovtunov AI, Iashkulov KB, Gromashevskii VL, Dzharkenov AF, Shchelkanov MI, Circulation of West Nile virus (Flaviviridae, Flavivirus) and some other arboviruses in the ecosystems of Volga delta, Volga-Akhtuba flood-lands and adjoining arid regions (2000-2002) [in Russian]. Vopr Virusol. 2004;49:45–51.PubMedGoogle Scholar
- Lvov DK, Butenko AM, Gromashevsky VL, Kovtunov AI, Prilipov AG, Kinney R, West Nile virus and other zoonotic viruses in Russia: examples of emerging-reemerging situations. Arch Virol Suppl. 2004;18:85–96.PubMedGoogle Scholar
- Kuno G, Chang GJ, Tsuchiya KR, Karabatsos N, Cropp CB. Phylogeny of the genus Flavivirus. J Virol. 1998;72:73–83.PubMedGoogle Scholar
- Burke DS, Monath TP. Flaviviruses. In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, et al. editors. Fields virology, vol. 1. Philadelphia: Lippincott Williams & Wilkins; 2001. p. 1055–109.
- Gaunt MW, Sall AA, de Lamballerie X, Falconar AK, Dzhivanian TI, Gould EA. Phylogenetic relationships of flaviviruses correlate with their epidemiology, disease association and biogeography. J Gen Virol. 2001;82:1867–76.PubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 11, Number 2—February 2005
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Norbert Nowotny, Zoonoses and Emerging Infections Group, Clinical Virology, Clinical Department of Diagnostic Imaging, Infectious Diseases and Clinical Pathology, University of Veterinary Medicine, Vienna, A-1210 Vienna, Austria; fax: 43 1 250772790
Top