Volume 12, Number 6—June 2006
Dispatch
Hantaviruses in Serbia and Montenegro
Figure 1
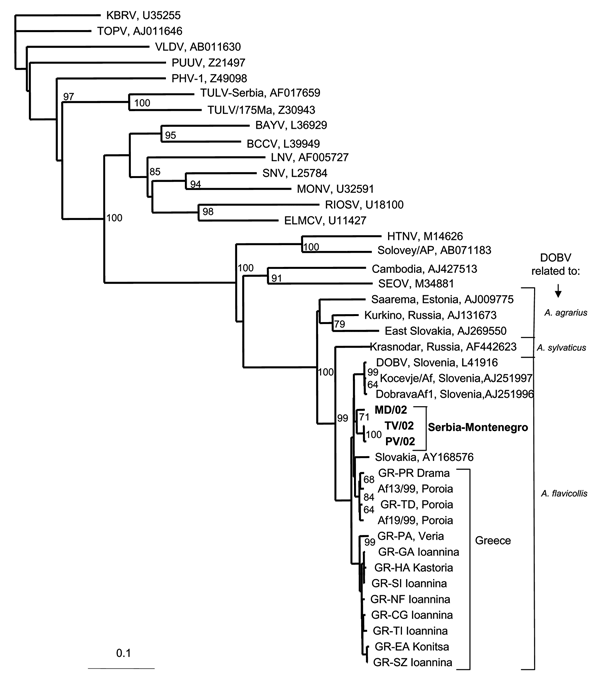
Figure 1. Phylogenetic tree based on partial S segment fragment showing the clustering of the sequence obtained from this study and respective representative hantavirus strains from GenBank database. The numbers indicate percentage bootstrap replicates (of 100); values <60% are not shown. Horizontal distances are proportional to the nucleotide differences. The scale bar indicates 10% nucleotide sequence divergence. Vertical distances are for clarity only. BAYV, Bayou virus; BCCV, Black Creek Canal virus; ELMCV, El Moro Canyon virus; HTNV, Hantaan virus; KBRV, Khabarovsk virus; LNV, Laguna Negra; MONV, Monongahela virus; NYV, New York virus; PHV, Prospect Hill virus; PUUV, Puumala virus; RIOSV, Rio Segundo virus; SEOV, Seoul virus; SNV, Sin Nombre virus; TOPV, Topografov virus; TULV, Tula virus; VLDV, Vladivostok virus. Accession numbers of Greek DOBV strains are AF060014–AF060024 for sequences from human cases and AJ410615 and AJ410619 from Apodemus flavicollis (Afl) sequences. Sequences in this study are indicated in boldface.