Volume 15, Number 9—September 2009
Research
Recent Ancestry of Kyasanur Forest Disease Virus
Rajeev Mehla, Sandeep R.P. Kumar, Pragya D. Yadav, Pradip V. Barde, Prasanna N. Yergolkar, Bobbie R. Erickson, Serena A. Carroll, Akhilesh C. Mishra, Stuart T. Nichol, and Devendra T. Mourya
Figure 2
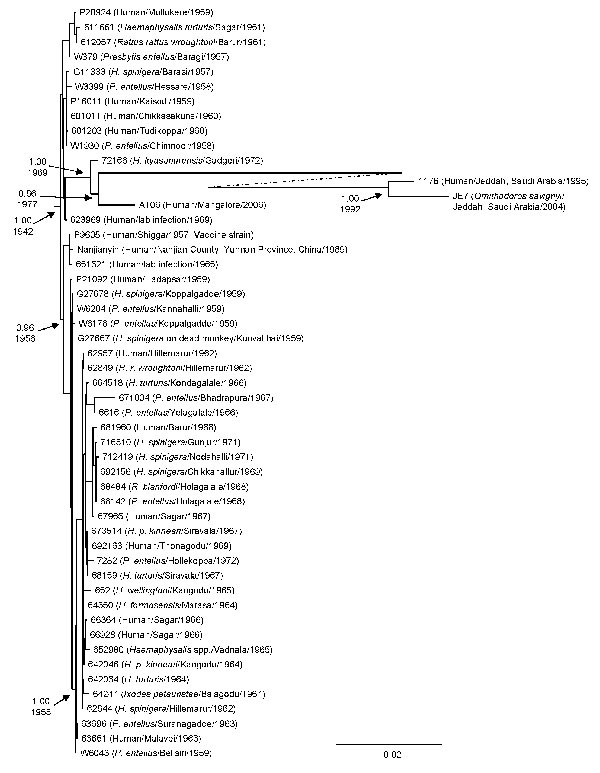
Figure 2. Bayesian coalescent analysis of sequence differences of Kyasanur Forest disease virus isolates from India (1957–2006), People’s Republic of China (1989), and Saudi Arabia (1995–2004). Analysis was conducted by using the general time reversible model incorporating invariant sites, a relaxed molecular clock, constant population size, and the BEAST, BEAUTi, and Tracer analysis software (26). The maximum clade credibility tree is depicted. Posterior probability values are indicated for clades of interest with the time to most recent common ancestor shown below. Scale bar indicates nucleotide substitutions per site.
References
- Thiel H-J, Collett MS, Gould EA, Heinz FX, Houghton M, Meyers G, Flaviviridae. In: Fauquet CM, Mayo MA, Maniloff J, Desslberger U, Ball LA, editors. Virus taxonomy: Eighth report of the International Committee on Taxonomy of Viruses. San Diego (CA): Elsevier Academic Press; 2005. p. 981–98.
- Banerjee K. Kyasanur Forest disease. In: Monath TP, editor. Arboviruses: epidemiology and ecology. Boca Raton (FL): CRC Press; 1990. p. 93–116.
- Bhatt PN, Work TH, Varma MGR, Trapido H, Narasimha Murthy DP, Rodrigues FM. Isolation of Kyasanur Forest disease from infected humans and monkeys of Shimoga District, Mysore State. Indian J Med Sci. 1966;20:316–20.PubMedGoogle Scholar
- Upadhyaya S, Narasimha Murthy DP, Anderson CR. Kyasanur forest disease in the human population of Shimoga district, Mysore state (1959–1966). Indian J Med Res. 1975;63:1556–63.PubMedGoogle Scholar
- Sreenivasan MA, Bhat HR, Rajagopalan PK. The epizootics of Kyasanur Forest disease in wild monkeys during 1964 to 1973. Trans R Soc Trop Med Hyg. 1986;80:810–4. DOIPubMedGoogle Scholar
- Bhat UK, Goverdhan MK. Transmission of Kyasanur Forest disease virus by the soft tick, Ornithodoros cross. Acta Virol. 1973;17:337–42.PubMedGoogle Scholar
- Bhat HR, Sreenivasan MA, Nayak SV. Susceptibility of common giant flying squirrel to experimental infection with KFD virus. Indian J Med Res. 1979;69:697–700.PubMedGoogle Scholar
- Goverdhan MK, Anderson CR. Reaction of Rattus rattus wroughtoni to Kyasanur forest disease virus. Indian J Med Res. 1978;67:5–10.PubMedGoogle Scholar
- Goverdhan MK, Anderson CR. The reaction of Funambulus tristriatus, Rattus blanfordi and Suncus murinus to Kyasanur forest disease virus. Indian J Med Res. 1981;74:141–6.PubMedGoogle Scholar
- Banerjee K, Bhat HR. Kyasanur forest disease. In: Mishra A, Polasa H, editors. Virus ecology. New Delhi (India): South Asian Publisher; 1984. p. 123–38.
- Zaki AM. Isolation of a flavivirus related to the tick-borne encephalitis complex from human cases in Saudi Arabia. Trans R Soc Trop Med Hyg. 1997;91:179–81. DOIPubMedGoogle Scholar
- Wang J, Zhang H, Fu S, Wang H, Ni D, Nasci R, Isolation of Kyasanur forest disease virus from febrile patient, Yunnan, China. Emerg Infect Dis. 2009;15:326–8. DOIPubMedGoogle Scholar
- Qattan I, Akbar N, Afif H, Abu Azmah S, Al-Khateeb T, Zaki A, A novel flavivirus: Makkah Region, 1994–1996. Saudi Epidemiology Bulletin. 1996;3:1–3.
- Charrel RN, Zaki AM, Attoui H, Fakeeh M, Billoir F, Yousef AI, Complete coding sequence of the Alkhurma virus, a tick-borne flavivirus causing severe hemorrhagic fever in humans in Saudi Arabia. Biochem Biophys Res Commun. 2001;287:455–61. DOIPubMedGoogle Scholar
- Zhang TS, Wang YM, Zhang YH, Duan S. A survey of antibodies to arboviruses in residents of southwestern Yunnan Province [in Chinese]. Chin J Endemiology. 1989;10:74–7.
- Hou ZL, Huang WL, Zi DY, Zhang HL, Shi HF. Study of the serologic epidemiology of tick-borne viruses in Yunnan [in Chinese]. Chinese Journal of Vector Biology and Control. 1992;3:173–6.
- Zanotto PM, Gao GF, Gritsun T, Marin MS, Jiang WR, Venugopal K, An arbovirus cline across the northern hemisphere. Virology. 1995;210:152–9. DOIPubMedGoogle Scholar
- Gould EA, de Lamballerie X, Zanotto PM, Holmes EC. Evolution, epidemiology, and dispersal of flaviviruses revealed by molecular phylogenies. Adv Virus Res. 2001;57:71–103. DOIPubMedGoogle Scholar
- Zanotto PM, Gould EA, Gao GF, Harvey PH, Holmes EC. Population dynamics of flaviviruses revealed by molecular phylogenies. Proc Natl Acad Sci U S A. 1996;93:548–53. DOIPubMedGoogle Scholar
- Katoh K, Kuma K, Toh H, Miyata T. MAFFT Version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005;33:511–8. DOIPubMedGoogle Scholar
- Galtier N, Gouy M, Gautier C. SeaView and Phylo_win, two graphic tools for sequence alignment and molecular phylogeny. Comput Appl Biosci. 1996;12:543–8.PubMedGoogle Scholar
- Farris JS, Kallersjo M, Kluge AG, Bult C. Testing significance of incongruence. Cladistics. 1994;10:315–9. DOIGoogle Scholar
- Swofford DL. PAUP*: phylogenetic analysis using parsimony (*and other methods), version 4.0b 10. Sunderland (MA): Sinauer Associates; 2003.
- Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–8. DOIPubMedGoogle Scholar
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. DOIPubMedGoogle Scholar
- Charrel RN, Zaki AM, Fakeeh M, Yousef AI, de Chesse R, Attoui H, Low diversity of Alkhurma hemorrhagic fever virus, Saudi Arabia, 1994–1999. Emerg Infect Dis. 2005;11:683–8.PubMedGoogle Scholar
- Grard G, Moureau G, Charrel RN, Lemasson J-J, Gonzalez J-P, Gallian P, Genetic characterization of tick-borne flaviviruses: new insights into evolution, pathogenetic determinants and taxonomy. Virology. 2007;361:80–92. DOIPubMedGoogle Scholar
- Charrel RN, Fagbo S, Moureau G, Alqahtani MH, Temmam S, de Lamballerie X. Alkhurma hemorrhagic fever virus in Ornithodoros savignyi ticks. Emerg Infect Dis. 2007;13:153–5. DOIPubMedGoogle Scholar
- Baillie GJ, Kolokotronis SO, Waltari E, Maffei JG, Kramer LD, Perkins SL. Phylogenetic and evolutionary analyses of St. Louis encephalitis virus genomes. Mol Phylogenet Evol. 2008;47:717–28. DOIPubMedGoogle Scholar
- Bryant JE, Holmes EC, Barrett AD. Out of Africa: a molecular perspective on the introduction of yellow fever virus into the Americas. PLoS Pathog. 2007;3:e75. DOIPubMedGoogle Scholar
- Boshell J. Kyasanur Forest disease: ecologic considerations. Am J Trop Med Hyg. 1969;18:67–80.PubMedGoogle Scholar
- Boshell JM, Rajagopalan PK. Observations on the experimental exposure to the monkeys, rodents and shrews to infestation of ticks in forest of Kyasanur forest disease area. Indian J Med Res. 1968;56:586–8.
- Ali S, Ripley SD. Handbook of the birds of India and Pakistan. Vol 1–10. New York: Oxford University Press; 2002.
Page created: December 07, 2010
Page updated: December 07, 2010
Page reviewed: December 07, 2010
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.