Volume 16, Number 6—June 2010
Dispatch
Novel Betaherpesvirus in Bats
Figure
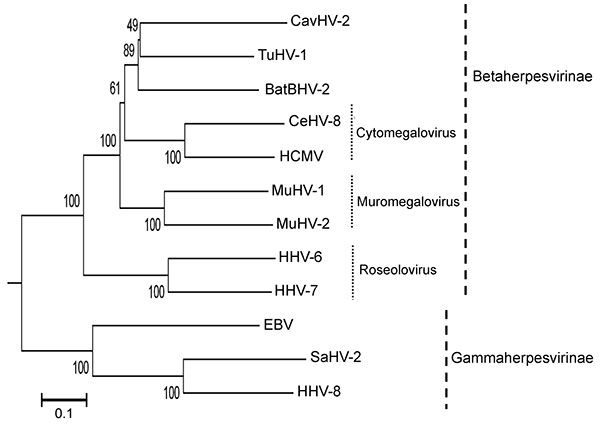
Figure. Phylogenetic tree based on the deduced amino acid sequences of complete glycoprotein B. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) is shown next to the branches. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The tree was rooted to herpes simplex virus type 1 (X14112). The evolutionary distances were computed by using the Poisson correction method and are in units of the number of amino acid substitutions per site. All positions containing gaps and missing data were eliminated from the dataset. The final dataset included a total of 698 positions. Phylogenetic analyses were conducted in MEGA4 (8). The herpesviruses used for comparison and their accession numbers are as follows: Epstein-Barr virus 1 (EBV, NC_007605), caviid herpesvirus (CavHV-2, FJ355434); mouse cytomegalovirus (MuHV-1, NC_004065), human cytomegalovirus (HCMV, X17403), human herpesvirus 6 (HHV-6, AF157706), human herpesvirus 7 (HHV-7, AF037218), human herpesvirus 8 (HHV-8, AF148805), rat cytomegalovirus (MuHV-2, NC_002512), cercopithecine herpesvirus 8 (CeHV-8, AY186194), saimiriine herpesvirus 2 (SaHV-2, NC_001350), and tupaiid herpesvirus 1 (TuHV-1, AF281817). Scale bar indicates evolutionary distance.
References
- Wong S, Lau S, Woo P, Yuen KY. Bats as a continuing source of emerging infections in humans. Rev Med Virol. 2007;17:67–91. DOIPubMedGoogle Scholar
- Maeda K, Hondo E, Terakawa J, Kiso Y, Nakaichi N, Endoh D, Isolation of novel adenovirus from fruit bat (Pteropus dasymallus yayeyamae). Emerg Infect Dis. 2008;14:347–9. DOIPubMedGoogle Scholar
- Omatsu T, Watanabe S, Akashi H, Yoshikawa Y. Biological characters of bats in relation to natural reservoir of emerging viruses. Comp Immunol Microbiol Infect Dis. 2007;30:357–74. DOIPubMedGoogle Scholar
- Watanabe S, Ueda N, Iha K, Masangkay JS, Fujii H, Alviola P, Detection of a new bat gammaherpesvirus in the Philippines. Virus Genes. 2009;39:90–3. DOIPubMedGoogle Scholar
- Watanabe S, Mizutani T, Sakai K, Kato K, Tohya Y, Fukushi S, Ligation-mediated amplification for effective rapid determination of viral RNA sequences (RDV). J Clin Virol. 2008;43:56–9. DOIPubMedGoogle Scholar
- Mizutani T, Endoh D, Okamoto M, Shirato K, Shimizu H, Arita M, Rapid genome sequencing of RNA viruses. Emerg Infect Dis. 2007;13:322–4. DOIPubMedGoogle Scholar
- VanDevanter DR, Warrener P, Bennett L, Schultz ER, Coulter S, Garber RL, Detection and analysis of diverse herpesviral species by consensus primer PCR. J Clin Microbiol. 1996;34:1666–71.PubMedGoogle Scholar
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–9. DOIPubMedGoogle Scholar
- Razafindratsimandresy R, Jeanmaire EM, Counor D, Vasconcelos PF, Sall AA, Reynes JM. Partial molecular characterization of alphaherpesviruses isolated from tropical bats. J Gen Virol. 2009;90:44–7. DOIPubMedGoogle Scholar
- Wibbelt G, Kurth A, Yasmum N, Bannert M, Nagel S, Nitsche A, Discovery of herpesviruses in bats. J Gen Virol. 2007;88:2651–5. DOIPubMedGoogle Scholar