Volume 16, Number 9—September 2010
Dispatch
KI and WU Polyomaviruses and CD4+ Cell Counts in HIV-1–infected Patients, Italy
Muhammed Babakir-Mina, Massimo Ciccozzi, Francesca Farchi, Massimiliano Bergallo, Rossana Cavallo, Gaspare Adorno, Carlo Federico Perno, and Marco Ciotti
Figure
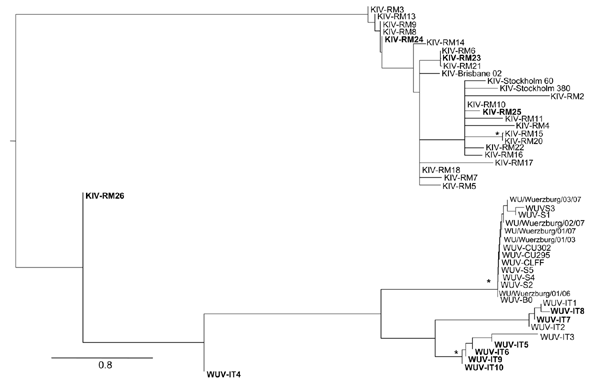
Figure. Maximum likelihood phylogenetic analysis of KI polyomavirus (KIPyV) and WU polyomavirus (WUPyV) small T antigen sequences. Strains identified in this study are in boldface. The tree was rooted by using the midpoint rooting method. Branch lengths were estimated by using the best fitting nucleotide substitution (Hasegawa, Kishino, and Yano) model according to a hierarchical likelihood ratio test (6,7) and were drawn to scale. Scale bar indicates 0.8 nt substitutions per site. Asterisks along the branches indicate significant statistical support for the clade subtending that branch (p<0.001 by the zero-branch-length test and bootstrap support >65%).
References
- Padgett BL, Walker DL, ZuRhein GM, Eckroade RJ, Dessel BH. Cultivation of papova-like virus from human brain with progressive multifocal leucoencephalopathy. Lancet. 1971;1:1257–60. DOIPubMedGoogle Scholar
- Gardner SD, Field AM, Coleman DV, Hulme B. New human papovavirus (BK) isolated from urine after renal transplantation. Lancet. 1971;1:1253–7. DOIPubMedGoogle Scholar
- Allander T, Andreasson K, Gupta S, Bjerkner M, Bogdanovic G, Persson MA, Identification of a third human polyomavirus. J Virol. 2007;81:4130–7. DOIPubMedGoogle Scholar
- Gaynor AM, Nissen MD, Whiley DM, McKay IM, Lambert SB, Wu G, Identification of a novel polyomavirus from patients with acute respiratory tract infections. PLoS Pathog. 2007;3:e64. DOIPubMedGoogle Scholar
- Sharp CP, Norja P, Anthony I, Bell JE, Simmonds P. Reactivation and mutation of newly discovered WU, KI, and Merkel cell carcinoma polyomaviruses in immunosuppressed individuals. J Infect Dis. 2009;199:398–404. DOIPubMedGoogle Scholar
- Babakir-Mina M, Ciccozzi M, Trento E, Perno CF, Ciotti M. KI and WU polyomaviruses in patients infected with HIV-1, Italy. Emerg Infect Dis. 2009;15:1323–5. DOIPubMedGoogle Scholar
- Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–8. DOIPubMedGoogle Scholar
- Babakir-Mina M, Ciccozzi M, Bonifacio D, Bergallo M, Costa C, Cavallo R, Identification of the novel KI and WU polyomaviruses in human tonsils. J Clin Virol. 2009;46:75–9. DOIPubMedGoogle Scholar
- Bergallo M, Terlizzi ME, Astegiano S, Ciotti M, Babakir-Mina M, Perno CF, Real time PCR TaqMan assays for detection of polyomaviruses KIV and WUV in clinical samples. J Virol Methods. 2009;162:69–74. DOIPubMedGoogle Scholar
- Babakir-Mina M, Ciccozzi M, Alteri C, Polchi P, Picardi A, Greco F, Excretion of the novel polyomaviruses KI and WU in the stool of patients with hematological disorders. J Med Virol. 2009;81:1668–73. DOIPubMedGoogle Scholar
- Venter M, Visser A, Lassauniere R. Human polyomaviruses, WU and KI in HIV exposed children with acute lower respiratory tract infections in hospitals in South Africa. J Clin Virol. 2009;44:230–4. DOIPubMedGoogle Scholar
- Mourez T, Bergeron A, Ribaud P, Scieux C, de Latour RP, Tazi A, Polyomaviruses KI and WU in immunocompromised patients with respiratory disease. Emerg Infect Dis. 2009;15:107–9. DOIPubMedGoogle Scholar
- Barzon L, Squarzon L, Militello V, Trevisan M, Porzionato A, Macchi V, WU and KI polyomaviruses in the brains of HIV-positive patients with and without progressive multifocal leukoencephalopathy. J Infect Dis. 2009;200:1755–8. DOIPubMedGoogle Scholar
- Kean JM, Rao S, Wang M, Garcea RL. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009;5:e1000363. DOIPubMedGoogle Scholar
- Dolei A, Pietropaolo V, Gomes E, Di Taranto C, Ziccheddu M, Spanu MA, Polyomavirus persistence in lymphocytes: prevalence in lymphocytes from blood donors and healthy personnel of a blood transfusion centre. J Gen Virol. 2000;81:1967–73.PubMedGoogle Scholar
Page created: August 28, 2011
Page updated: August 28, 2011
Page reviewed: August 28, 2011
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.