Chikungunya Virus, Southeastern France
Marc Grandadam

, Valérie Caro, Sébastien Plumet, Jean-Michel Thiberge, Yvan Souarès, Anna-Bella Failloux, Hugues J. Tolou, Michel Budelot, Didier Cosserat, Isabelle Leparc-Goffart, and Philippe Desprès
Author affiliations: Author affiliations: Institut Pasteur, Paris, France (M. Grandadam, V. Caro, J.-M. Thiberge, A.-B. Failloux, P. Desprès); Institut de Recherche Biomédicale des Armées, Marseille, France (S. Plumet, H.J. Tolou, I. Leparc-Goffart); Institut de Veille Sanitaire, Saint-Maurice, France (Y. Souarès); Private Consulting Rooms, Saint-Raphaël, France (M. Budelot, D. Cosserat)
Main Article
Figure
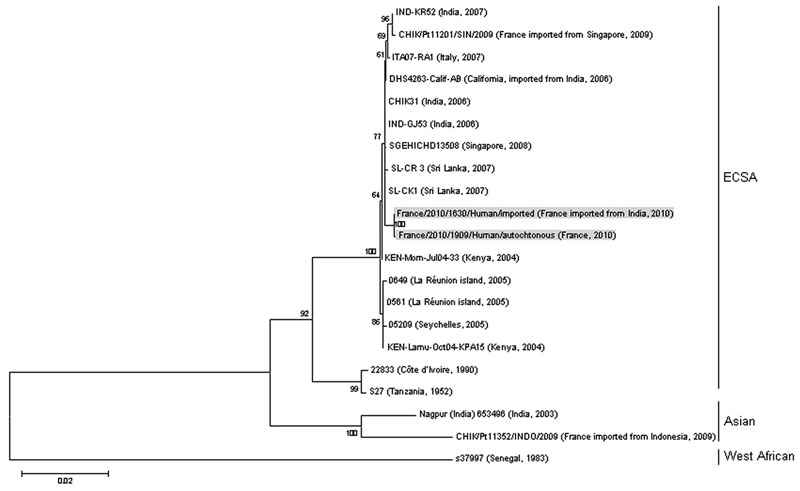
Figure. Phylogenetic relationships among chikungunya virus isolates from cases of chikungunya fever in France, based on complete E2-6K-E1 nucleotide sequence (2,771 nt) analysis. Gray shading indicates imported and autochthonous strains. Sequence alignments were performed by using BioNumerics version 5.1 (www.applied-maths.com). Phylogenetic analysis was inferred by using the maximum-likelihood method as implemented in MEGA version 5 software (www.megasoftware.net). Bootstrap support values (1,000 replicates) are indicated at major nodes. The sequence of the strains from France described in this study has been deposited in GenBank (accession number pending); other sequences were retrieved from GenBank. Scale bar indicates number of base substitutions per site. ECSA, east/central/south Asia.
Main Article
Page created: August 14, 2011
Page updated: August 14, 2011
Page reviewed: August 14, 2011
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.