Volume 17, Number 9—September 2011
Research
Classical Bovine Spongiform Encephalopathy by Transmission of H-Type Prion in Homologous Prion Protein Context
Abstract
Bovine spongiform encephalopathy (BSE) and BSE-related disorders have been associated with a single major prion strain. Recently, 2 atypical, presumably sporadic forms of BSE have been associated with 2 distinct prion strains that are characterized mainly by distinct Western blot profiles of abnormal protease-resistant prion protein (PrPres), named high-type (BSE-H) and low-type (BSE-L), that also differed from classical BSE. We characterized 5 atypical BSE-H isolates by analyzing their molecular and neuropathologic properties during transmission in transgenic mice expressing homologous bovine prion protein. Unexpectedly, in several inoculated animals, strain features emerged that were highly similar to those of classical BSE agent. These findings demonstrate the capability of an atypical bovine prion to acquire classical BSE–like properties during propagation in a homologous bovine prion protein context and support the view that the epidemic BSE agent could have originated from such a cattle prion.
Transmissible spongiform encephalopathies, or prion diseases, are a group of neurodegenerative disorders that include Creutzfeldt-Jakob disease (CJD) in humans, scrapie in sheep and goats, and bovine spongiform encephalopaty (BSE) in cattle. Prion diseases are characterized by specific histopathologic lesions and deposits of an abnormal conformational isoform (PrPSc) of the host-encoded physiologic prion protein (PrPC) in the central nervous system. PrPSc but not PrPC is partially resistant to digestion by proteinase K, resulting in an N terminally truncated prion protein termed PrPres that can be detected by Western blot and showing a characteristic banding pattern that reflects the 3 PrPres glycoforms. The apparent molecular masses and relative quantities of these glycoforms are used in biochemical PrPres typing as the criteria to differentiate between prion diseases.
BSE is a prion epidemic that has caused the deaths of ≈200,000 cattle in Europe, mainly in the United Kingdom, since it emerged in 1985. Although multiple agent strains have been identified in sheep scrapie (1,2) and human CJD (3,4), early evidence showed that BSE was caused by a single major strain (5,6) with the ability to efficiently cross the species barriers and showing stable features even when transmitted to other species. Transmission of BSE to humans through contaminated food is believed to be responsible for variant CJD (vCJD) (7,8). Several authors reported that BSE and vCJD prions share similar strain-specific features, including a unique PrPres molecular signature (6,9,10), after transmission to mice or macaques. However, other studies described the production of different PrPres molecular signature after BSE and vCJD prions transmission in wild-type (11) and human PrP transgenic mice (12,13).
Epidemiologic investigations identified contaminated meat and bone meal as the vehicle that recycled the BSE agent in the cattle population (14). However, the origin of BSE remains under debate, and the disease has been hypothesized to have derived either from sheep scrapie or from a spontaneous bovine prion disease analogous to sporadic forms of CJD in human (15) or even from human transmissible spongiform encephalopathy (16).
More recently, 2 atypical forms of BSE have been identified in several European countries (17), Japan (18,19), the United States (20), and Canada (21). Several studies suggest that these atypical disorders are associated with 2 distinct prion strains that are mainly characterized by distinct PrPres profiles, named high-type (H-type) and low-type (L-type) according to the electrophoretic migration of the unglycosylated PrPres, which is higher (BSE-H) or lower (BSE-L) than classical BSE (BSE-C) (22). An additional distinctive signature of H-type and L-type PrPres is the smaller proportion of the diglycosylated PrPres compared with the classical-type (C-type) PrPres, more obvious in L-type BSE (23–25).
All epidemiologic and biologic evidence strongly suggests that BSE-H and BSE-L represent sporadic forms of BSE (23,24) associated with 2 distinct prion strains. Transmission experiments in different mouse models, including transgenic mice expressing bovine PrP, showed that BSE-H and BSE-L exhibited strain-specific features clearly distinct between each other that also differed from BSE-C (13,25–28). However, BSE-L isolates unexpectedly showed transmission of a disease with some phenotypic features that resembled those of the BSE-C agent when inoculated in either transgenic mice expressing ovine PrP (28) or inbred wild-type mouse lines (25), suggesting that atypical bovine strains can modify their properties, at least after species barrier passages, converging with those of BSE-C.
We show that the transmission of atypical BSE-H isolates in transgenic mice expressing homologous bovine prion protein (PrP) led to emergence of a clearly distinct prion with strain features similar to those of the BSE-C agent and that such similarities were maintained on subsequent passages. These observations provide new insights into the nature of the events that could have led to the BSE epizootic.
Transgenic Mice
We used Tg110 transgenic mice in all inoculation experiments. This mouse line expresses bovine PrPC (≈8× that of the level of PrPC in cattle brain) under the control of the mouse prnp gene promoter in a mouse PrP0/0 background (29).
BSE Isolates
The 5 BSE-H isolates used in this study comprised brainstem samples from naturally affected cows, diagnosed as atypical H-type BSE on the basis of the molecular analyses of PrPres (23). All cows were healthy and killed at 8–15 years of age. Four (isolates 07-644, 03-440, 03-2095, and 02-2695) were provided by the Agence Française de Sécurité Sanitaire des Aliments (Lyon, France). Isolate 45 was obtained from the Polish National Veterinary Research Institute (Pulawy, Poland). For comparative studies, material obtained from brainstem of 1 cow naturally infected with BSE-C (RQ 225:PG1199/00), supplied by the Veterinary Laboratories Agency (New Haw, Addlestone, Surrey, UK), was used as BSE-C control. For mouse inoculations, all isolates were prepared from brain tissues as 10% (wt/vol) homogenates. For subpassages, 10% brain homogenates from Tg110 mice collected from primary passage were used as inocula.
All inocula were prepared in sterile 5% glucose as 10% homogenates. Each inoculum was prepared separately in a biosafety cabinet according to a strict protocol to avoid cross-contamination. To diminish the risk for bacterial infection, the inocula were preheated for 10 min at 70°C before inoculation.
Mouse Transmission Studies
Groups of 6–12 mice (6–7 weeks of age, weighing ≈20 g) were inoculated with 20 µL of the appropriate sample in the right parietal lobe by using 25-gauge disposable hypodermic syringes. UNO MICRO ID-8 ISO transponders (Roestvaststaal BV, Zevenaar, the Netherlands) were used for individual identification of mice. After inoculation, mice were observed daily and their neurologic status were assessed 2×/wk. When progression of the disease was evident, animals were euthanized. All animals were housed in accordance with guidelines of the Code for Methods and Welfare Considerations in Behavioral Research with Animals of the European Union directive 86/609EC). Necropsy was performed, and brain and spleen were taken. Part of the sample was frozen at −20°C for biochemical analysis, and the remaining part was fixed for histopathologic studies.
Survival times were calculated for each inoculum as the time between inoculation and euthanasia in days and expressed as the mean of the survival days postinoculation (dpi) of all the inoculated mice with its correspondent standard error of the mean. Data were processed by using SigmaPlot 2001 software (Systat Software, San Jose, CA, USA).
PrPres Western Blotting
Frozen mouse brain samples were prepared as 10% (wt/vol) homogenates in 5% glucose in distilled water in grinding tubes (Bio-Rad, Hercules, CA, USA) by using a TeSeE Precess 48 homogenizer (Bio-Rad) following the manufacturer’s instructions. All samples were analyzed by Western blot by using the kit TeSeE Western Blot 355 1169 (Bio-Rad) but with some adjustments for the different amounts of samples used. To achieve the volume proposed in the manufacturer’s recommendations, 100 μL of the brain homogenates to be tested were supplemented with 100 μL of a 10% brain homogenate from PrP null mice (30). Processed samples were loaded on Criterion 12% polyacrylamide gels from Bio-Rad (165.6001) and electrotransferred to immobilon membranes (IPVH 000 10 [Millipore, Billerica, MA, USA]). For the immunoblotting experiments, Sha31 (31), Saf84 (Cayman Chemical, Ann Arbor, MI, USA) and 12B2 (32) monoclonal antibodies (mAbs) were used at a concentration of 1 μg/mL. Sha31 recognizes 156YEDRYYRE163 epitope, and Saf84 recognizes 171QVYYRPVDQYS181 epitope and 12B2 recognizes 101WGQGG105 epitope of the bovine PrP sequence. Immunocomplexes were detected by horseradish peroxidase–conjugated antimouse immunoglobulin G (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Immunoreactivity was visualized by chemiluminescence (Amersham Pharmacia Biotech) and obtained after exposition with medical radiographic film (Agfa, Mortsel, Belgium).
Histopathologic Analysis
All procedures involving mouse brains and spleens were performed as described (33). Briefly, samples were fixed in neutral-buffered 10% formalin (4% formaldehyde) before embedding in paraffin. Once deparaffinized, 2-µm–thick tissue sections were stained with hematoxylin and eosin. Lesion profiles of the brains were established according to the standard method described by Fraser and Dickinson (34). For paraffin-embedded tissue blots, the protocol described by Andréoletti et al. (35) was used.
Figure 1
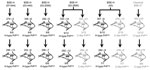
Figure 1. Overview of transmission of BSE-H isolates in tg110 mice. Five different isolates were intracerebrally inoculated into groups of 6–12 mice per isolate. Survival times at different serial passages are indicated as...
The transmission dynamic of BSE-H agent into Tg110 mice is similar to that of BSE-C. The 4 BSE-H isolates from France and 1 from Poland that were intracerebrally inoculated into transgenic mice expressing bovine PrP (Tg110 mice) induced a typical neurologic disease on primary transmission, with a 100% attack rate (Figure 1). Remarkably, the survival times (mean ± SD 274 ± 3 to 346 ± 6 dpi) were similar than those produced by several BSE-C isolates (≈300 days) on the same Tg110 mouse line (29,36,37). The longest mean survival times observed for mice infected with isolates 03–440 (mean ±SD 346 ±6 dpi) and 02–2695 (330 ±14 dpi) could reflect a lower infectivity of these isolates, consistent with its comparatively lower PrPres content (data not shown). Moreover, the survival time of mice infected with these 2 isolates was reduced on subpassage, approaching that for BSE-C or BSE-H isolates of presumably higher titer (i.e., producing no substantial reduction of survival time on subpassage: isolates 07-644, 03-2095, and 45).
PrPres Molecular Profiles of BSE-H–Inoculated Tg110 Mice
Figure 2
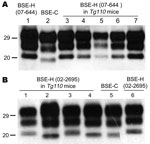
Figure 2. Western blot analyses of brain protease-resistant prion protein (PrPres) from BSE-H infected mice by using Sha31 monoclonal antibody. A) Mice infected with isolate 07-644 at first passage (lanes 3–7) showing a...
The brains of inoculated mice were examined for PrPres by Western blot analysis with Sha31 mAb. Consistent with the efficient transmission observed, PrPres was readily detected from the first passage in all Tg110 mice inoculated with the different BSE-H isolates. In 3 BSE-H isolates (07-644, 03-440, and 03-2095), the totality (100%) of the inoculated Tg110 mice produced a PrPres profile similar to that in cattle (H-type PrPres) but clearly distinct from that produced by BSE-C agent (C-type PrPres) in cattle (Figure 2, panel A; data not shown). Compared with C-type PrPres, H-type PrPres was characterized by a significantly higher apparent molecular mass of the unglycosylated band. The results obtained with these 3 isolates were comparable with those obtained by other authors with 2 BSE-H isolates inoculated in a different bovine PrP mouse line (27), where they concluded that the H-type agent essentially retained its biochemical phenotype upon serial transmission to bovine PrP transgenic mice.
Figure 3
Figure 3. Comparative Western blot analyses with Sha31 and 12B2 monoclonal antibodies (mAbs) of brain protease-resistant prion protein (PrPres) from BSE-H–infected mice. Mice infected with isolate 07-644 (lane 1), 02-2695 (lanes 2 and...
Figure 4
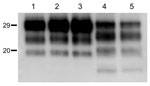
Figure 4. Western blot analyses of brain protease-resistant prion protein (PrPres) from BSE-H infected mice by using Saf84 monoclonal antibody. Tg110 mice infected with isolate 02-2695 (lanes 2 and 3) or 45 (lane...
We observed a different situation for the other 2 BSE-H isolates (02-2695 from France and 45 from Poland), where 3 and 2, respectively, of infected mice (Figure 1) showed a PrPres profile clearly distinct from that of BSE-H in cattle (Figure 2, panel B). These 5 mice produced a PrPres with lower (≈1.5 kDa) apparent molecular mass of the 3 PrPres glycoforms, which was indistinguishable from that produced by BSE-C agent in these mice (Figure 2, panel B, and data not shown). Further characterization of this PrPres with other antibodies showed that the PrPres produced by these mice was not recognized by 12B2 mAb (Figure 3). However, 12B2 immunoreactivity against the H-type PrPres produced by other mice (inoculated at the same time with the same isolates) remains essentially similar to that in cattle BSE-H (Figure 3). Furthermore, PrPres immunolabeling with Saf84 mAb showed that these mice, contrary to mice with H-type PrPres, did not retain the characteristic PrPres band profile (4 bands) of cattle BSE-H but showed a PrPres profile (3 bands) similar to that of BSE-C in Tg110 mice (Figure 4).
Figure 5
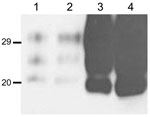
Figure 5. Comparison of the amount of protease-resistant prion protein (PrPres) in brain sample from mouse inoculated with BSE-H (isolate 02-2695) showing either high-type (lane 1 and 2, first and second passages, respectively)...
In addition, the proportion of the diglycosylated PrPres increased in comparison with the mice with H-type features, as shown by using Sha31 mAb (Figure 2, Figure 3). Another difference was that the PrPres level in brain was much higher than that in mouse brains with H-type PrPres but similar to that in mouse brains inoculated with BSE-C, as shown by comparative Western blot analysis by using Sha31 mAb (Figure 5) and by the different equivalent brain tissue masses loaded to obtain equivalent PrPres signals (Figure 2, Figure 3, Figure 4). A 10-fold equivalent brain tissue mass was loaded for brains from mice showing H-type PrPres molecular profile than from those with a classical-like (C-like) PrPres molecular profile to obtain equivalent PrPres signals (Figure 2, Figure 3, Figure 4). These mice thus showed PrPres molecular features indistinguishable from those in Tg110 mice infected with C-like features.
For these 2 isolates, a second passage was performed in Tg110 mice by using a brain homogenate derived from a mouse with either H-type or C-like PrPres (Figure 1). Survival times did not differ substantially when the different inocula were compared (H-type vs. C-like PrPres brain homogenate) or when compared with second passages of BSE-C in these mice. All the mice inoculated with the H-type brain homogenates showed H-type PrPres features, whereas all the mice inoculated with the C-like brain homogenates exhibited C-type PrPres molecular features indistinguishable from that of BSE-C (data not shown).
Lesion Profiles and PrPSc Deposition Patterns in BoPrP-Tg110 Mice
Figure 6

Figure 6. Vacuolar lesion profiles in brains from Tg110 mice inoculated with BSE-H (isolate 02-2695, first passage) showing either H-type PrPres phenotype (black triangles, n = 6 animals) or C-like PrPres phenotype (blue...
We next examined vacuolation and PrPSc distribution in the brain, which are known to vary by strain (10,34,38). In general, mice brains exhibiting H-type PrPres correlated with overall more intensive vacuolation that is pronounced in areas such as the hypothalamus, medial thalamus, and mesencephalic tegmentum (Figure 6). However, a different situation was observed when we studied the brains of BSE-H–infected mice exhibiting C-like PrPres features. All these mice showed a lesion pattern comparable with that in BSE-C–infected mice, in which slight differences are found only in the mesencephalic tegmentum (Figure 6). These differences consisted of moderate lesions, whereas in BSE-C–infected mice, no lesions were found in this area. These features were conserved on secondary transmissions where no remarkable differences were found when compared with primary transmissions (data not shown).
Figure 7
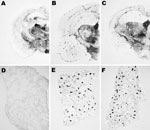
Figure 7. Abnormal isoform of host-encoded prion protein (PrPSc) deposition patterns in brain and spleen from Tg110 mice infected with BSE-H. A–C) Paraffin-embedded tissue (PET) blots of representative coronal sections at the level...
Moreover, PrPSc deposits were distinctly distributed after both primary and secondary transmissions when BSE-H–infected mice exhibiting C-type PrPres features were compared with those with H-type PrPres, as assessed by paraffin-embedded tissue blot on brain coronal sections (Figure 7). However, the PrPSc deposition patterns were clearly similar when these mice were compared with those infected with BSE-C, after both primary and secondary transmission.
Transmission experiments (27) showed that, contrary to BSE-C, BSE-H is poorly lymphotropic in mouse models. The comparative study of PrPSc accumulation in spleen from our Tg110 mice infected with both agents showed that BSE-H infected mice exhibiting H-type PrPres in their brain were consistently scored as negative for PrPSc detection by paraffin-embedded tissue blot. In contrast, clear PrPSc deposits were always detected in BSE-H–infected mice exhibiting C-like features, as in mice infected with BSE-C (Figure 7). Similar results were obtained after secondary transmissions.
We studied the behavior and stability of the atypical BSE-H during propagation into a bovine PrP background, thus in the absence of a species barrier. We used Tg110 mice (29,36) because they express a PrPC homologous to that of the donors, thus providing a relevant context for comparing atypical BSE-H and epizootic BSE-C isolates.
Our results showed that all BSE-H isolates induced a typical neurologic disease on primary transmission, with a 100% attack rate and survival times similar to those produced by several BSE-C isolates in this mouse line (29,36) (Figure 1). The longer survival times for some mice infected with BSE-H isolates could reflect a lower infectivity of this isolate consistent with the reduction of survival time observed on subpassages, approaching that for BSE-C or BSE-H isolates of presumably higher titer (i.e., producing no substantial reduction of survival time on subpassage). These results are also consistent with another comparative study of BSE-H and BSE-C transmissions in a different bovine PrP mouse line (27). These data suggest that atypical BSE-H and BSE-C agents have similar transmission features into Tg110 mice.
Although all BSE-H–inoculated mice showed homogeneous survival times, a phenotypic divergence was observed in a few animals infected with 2 of the BSE-H isolates. Surprisingly, these few mice showed phenotypic features clearly distinct from those in most of the BSE-H–infected mice but similar to those of BSE-C propagated onto the same mice, according to various criteria. First, a PrPres profile indistinguishable from that produced by BSE-C agent in these mice but clearly distinct from that of BSE-H in cattle, in terms of 1) apparent molecular mass of PrPres, 2) PrPres glycosylation pattern, 3) immunoreactivity with 12B2 mAb, and 4) pattern of labeling with Saf84 antibody. Second, the vacuolation profile essentially overlapped that in mice infected with BSE-C, with slight differences only in the mesencephalic tegmentum area. Third, the spatial distribution of PrPres in the brain was clearly similar to that of mice infected with BSE-C. Fourth, PrPSc was consistently detected in the spleen, similar to mice infected with BSE-C. These similarities with BSE-C were fully retained after a second passage by using brain homogenate from mice with C-like features, whereas a BSE-H strain phenotype was maintained in mice inoculated with mouse brains homogenates containing H-type PrPres.
However, C-like features emerged in only 2 of the 5 isolates tested. Because only a low proportion of the mice inoculated with these 2 isolates exhibited these novel features (3/12 and 2/10, respectively), the lack of such observation in the other 3 isolates, and in 2 other independent studies of 3 BSE-H isolates in different bovine transgenic mouse lines (27), could be due to the low number of inoculated mice (6 per isolate), which could be statistically insufficient for such an event. No variability was ever observed in the PrPres profiles of >100 Tg110 mice inoculated with 4 different BSE-C isolates (29,36) (Figure 1). However, a divergent evolution of the BSE agent has been reported after trans-species transmission in both wild-type (11) and human PrP transgenic mice (12,39,40).
Although further studies are required to clarify the mechanisms associated with the emergence of distinct phenotypes among individual mice, several factors would be expected to influence the probability of detecting such a variant through mouse bioassay. These factors are 1) amount or regions of cattle brain tissue taken for inoculum preparation, 2) physicochemical treatment during inoculum preparation (e.g., temperature, homogenization buffer), 3) the precise site of mouse inoculation, 4) the infectious titer of the inoculum, and 5) others unknown mouse factor affecting prion propagation and disease evolution. Because samples used in this study were prepared from the same region (brainstem) following the same precise protocol and under identical conditions, differences in inoculum preparation and conditions are unlikely. However, the possibility that the observations might be influenced by the precise neuroanatomic origin of the inoculated bovine brainstem homogenate or by other mouse bioassay–related factors cannot be excluded.
The possible cross-contamination of the BSE-H isolates material (02-2695 and 45 from 2 laboratories in different countries) by a BSE-C infectious source was judged highly improbable for several reasons. These reasons are 1) the strict biosafety procedures followed for sample collection, preparation of the inocula, inoculation scheme, and care of mice; 2) the absence of C-type PrPSc in the BSE-H inocula used for transmissions as deduced by Western blot analysis; and 3) 2 independent transmission experiments, involving separate batches of both incriminated isolates, all produced consistent results.
Together, these observations support 2 possible hypotheses. First, a minor strain component might be present in BSE-H isolates that could emerge on subsequent transmission in Tg110 mice. Second, a new strain component has been generated during propagation of BSE-H agent in Tg110. In both instances, emergence of the new strain, either in the original cattle or during propagation in Tg110 mice, could be promoted by specific propagation conditions or by physicochemical treatment of the inoculum. In this regard, acquisition of novel properties by a sporadic cattle transmissible spongiform encephalopathy agent by a physicochemical treatment, such as that applied to carcass-derived products, has been invoked as a possible origin for the BSE epidemic (7).
Contrary to BSE-H, the atypical BSE-L agent retained unique and distinct phenotypic features, compared with BSE-C agent, on transmission to both bovine and human PrP transgenic mice (26–28). This agent, however, acquired phenotypic traits intriguingly similar to those of the BSE agent during trans-species transmission in either transgenic mice expressing ovine PrP (28) or inbred mouse lines. On the basis of these observations, the BSE-C agent already has been speculated to have originated from atypical BSE-L after conversion in an intermediate host such as a sheep. However, the capacity of these BSE-L–derived agents to retain BSE phenotypic traits after reinoculation to bovine PrP transgenic mice is a key question, remaining to be demonstrated, to show whether the observed convergence truly reflects a permanent strain shift of the BSE-L agent rather than a phenotypic convergence in an experimental model.
In contrast, our results suggest that prion strain divergence might occur on propagation of atypical BSE-H in a homologous bovine PrP context and that this strain divergence could result from a permanent strain shift of the BSE-H agent toward a C-like agent that is stable in subsequent passages. These findings emphasize the potential capacity of prion diversification during propagation, even in the absence of any species barrier, and represent an experimental demonstration of the capability of an atypical, presumably sporadic, bovine prion to acquire C-like properties during propagation in a homologous bovine PrP context.
Results in transgenic mouse models cannot be directly extrapolated to the natural host. However, our observations are consistent with the view that the BSE agent could have originated from a cattle prion, such as BSE-H, and provide new insights into the nature of the events that could have led to the appearance of this agent.
Dr Torres is the lead researcher scientist of the Prions Group at the Centro de Investigación en Sanidad Animal–Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria, Madrid, Spain. His research interests include prion strain characterization and evolution and the pathogenesis of prion diseases and their effects on human and animal health.
Acknowledgment
This study was supported by grants from the European Union (CT2004-50657 and CT2004-023183) and from UK Food Standards Agency (M03043).
References
- Dickinson AG, Meikle VM. Host-genotype and agent effects in scrapie incubation: change in allelic interaction with different strains of agent. Mol Gen Genet. 1971;112:73–9. DOIPubMedGoogle Scholar
- Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274:2079–82. DOIPubMedGoogle Scholar
- Hill AF, Desbruslais M, Joiner S, Sidle KC, Gowland I, Collinge J, The same prion strain causes vCJD and BSE. Nature. 1997;389:448–50, 526. DOIPubMedGoogle Scholar
- Bruce M, Chree A, McConnell I, Foster J, Pearson G, Fraser H. Transmission of bovine spongiform encephalopathy and scrapie to mice: strain variation and the species barrier. Philos Trans R Soc Lond B Biol Sci. 1994;343:405–11. DOIPubMedGoogle Scholar
- Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, Transmissions to mice indicate that “new variant” CJD is caused by the BSE agent. Nature. 1997;389:498–501. DOIPubMedGoogle Scholar
- Aguzzi A, Glatzel M. Prion infections, blood and transfusions. Nat Clin Pract Neurol. 2006;2:321–9. DOIPubMedGoogle Scholar
- Lasmézas CI, Deslys JP, Demaimay R, Adjou KT, Lamoury F, Dormont D, BSE transmission to macaques. Nature. 1996;381:743–4. DOIPubMedGoogle Scholar
- Scott MR, Will R, Ironside J, Nguyen HO, Tremblay P, DeArmond SJ, Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc Natl Acad Sci U S A. 1999;96:15137–42. DOIPubMedGoogle Scholar
- Ritchie DL, Boyle A, McConnell I, Head MW, Ironside JW, Bruce ME. Transmissions of variant Creutzfeldt-Jakob disease from brain and lymphoreticular tissue show uniform and conserved bovine spongiform encephalopathy–related phenotypic properties on primary and secondary passage in wild-type mice. J Gen Virol. 2009;90:3075–82. DOIPubMedGoogle Scholar
- Asante EA, Linehan JM, Desbruslais M, Joiner S, Gowland I, Wood AL, BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J. 2002;21:6358–66. DOIPubMedGoogle Scholar
- Béringue V, Herzog L, Reine F, Le Dur A, Casalone C, Vilotte JL, Transmission of atypical bovine prions to mice transgenic for human prion protein. Emerg Infect Dis. 2008;14:1898–901. DOIPubMedGoogle Scholar
- Wilesmith JW, Wells GA. Bovine spongiform encephalopathy. Curr Top Microbiol Immunol. 1991;172:21–38.PubMedGoogle Scholar
- Colchester AC, Colchester NT. The origin of bovine spongiform encephalopathy: the human prion disease hypothesis. Lancet. 2005;366:856–61. DOIPubMedGoogle Scholar
- Jacobs JG, Langeveld JP, Biacabe AG, Acutis PL, Polak MP, Gavier-Widen D, Molecular discrimination of atypical bovine spongiform encephalopathy strains from a geographical region spanning a wide area in Europe. J Clin Microbiol. 2007;45:1821–9. DOIPubMedGoogle Scholar
- Yamakawa Y, Hagiwara K, Nohtomi K, Nakamura Y, Nishijima M, Higuchi Y, Atypical proteinase K–resistant prion protein (PrPres) observed in an apparently healthy 23-month-old Holstein steer. Jpn J Infect Dis. 2003;56:221–2.PubMedGoogle Scholar
- Masujin K, Shu Y, Yamakawa Y, Hagiwara K, Sata T, Matsuura Y, Biological and biochemical characterization of L-type-like bovine spongiform encephalopathy (BSE) detected in Japanese black beef cattle. Prion. 2008;2:123–8. DOIPubMedGoogle Scholar
- Richt JA, Kunkle RA, Alt D, Nicholson EM, Hamir AN, Czub S, Identification and characterization of two bovine spongiform encephalopathy cases diagnosed in the United States. J Vet Diagn Invest. 2007;19:142–54. DOIPubMedGoogle Scholar
- Dudas S, Yang J, Graham C, Czub M, McAllister TA, Coulthart MB, Molecular, biochemical and genetic characteristics of BSE in Canada. PLoS ONE. 2010;5:e10638. DOIPubMedGoogle Scholar
- Buschmann A, Biacabe AG, Ziegler U, Bencsik A, Madec JY, Erhardt G, Atypical scrapie cases in Germany and France are identified by discrepant reaction patterns in BSE rapid tests. J Virol Methods. 2004;117:27–36. DOIPubMedGoogle Scholar
- Biacabe AG, Laplanche JL, Ryder S, Baron T. Distinct molecular phenotypes in bovine prion diseases. EMBO Rep. 2004;5:110–5. DOIPubMedGoogle Scholar
- Casalone C, Zanusso G, Acutis P, Ferrari S, Capucci L, Tagliavini F, Identification of a second bovine amyloidotic spongiform encephalopathy: molecular similarities with sporadic Creutzfeldt-Jakob disease. Proc Natl Acad Sci U S A. 2004;101:3065–70. DOIPubMedGoogle Scholar
- Capobianco R, Casalone C, Suardi S, Mangieri M, Miccolo C, Limido L, Conversion of the BASE prion strain into the BSE strain: the origin of BSE? PLoS Pathog. 2007;3:e31. DOIPubMedGoogle Scholar
- Buschmann A, Gretzschel A, Biacabe AG, Schiebel K, Corona C, Hoffmann C, Atypical BSE in Germany—proof of transmissibility and biochemical characterization. Vet Microbiol. 2006;117:103–16. DOIPubMedGoogle Scholar
- Béringue V, Bencsik A, Le Dur A, Reine F, Lai TL, Chenais N, Isolation from cattle of a prion strain distinct from that causing bovine spongiform encephalopathy. PLoS Pathog. 2006;2:e112. DOIPubMedGoogle Scholar
- Béringue V, Andréoletti O, Le Dur A, Essalmani R, Vilotte JL, Lacroux C, A bovine prion acquires an epidemic bovine spongiform encephalopathy strain-like phenotype on interspecies transmission. J Neurosci. 2007;27:6965–71. DOIPubMedGoogle Scholar
- Castilla J, Gutiérrez-Adán A, Brun A, Pintado B, Ramirez MA, Parra B, Early detection of PrPres in BSE-infected bovine PrP transgenic mice. Arch Virol. 2003;148:677–91. DOIPubMedGoogle Scholar
- Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol. 1994;8:121–7. DOIPubMedGoogle Scholar
- Feraudet C, Morel N, Simon S, Volland H, Frobert Y, Creminon C, Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. J Biol Chem. 2005;280:11247–58. DOIPubMedGoogle Scholar
- Yull HM, Ritchie DL, Langeveld JP, van Zijderveld FG, Bruce ME, Ironside JW, Detection of type 1 prion protein in variant Creutzfeldt-Jakob disease. Am J Pathol. 2006;168:151–7. DOIPubMedGoogle Scholar
- Andréoletti O, Lacroux C, Chabert A, Monnereau L, Tabouret G, Lantier F, PrPSc accumulation in placentas of ewes exposed to natural scrapie: influence of foetal PrP genotype and effect on ewe-to-lamb transmission. J Gen Virol. 2002;83:2607–16.PubMedGoogle Scholar
- Fraser H, Dickinson AG. The sequential development of the brain lesion of scrapie in three strains of mice. J Comp Pathol. 1968;78:301–11. DOIPubMedGoogle Scholar
- Andréoletti O, Simon S, Lacroux C, Morel N, Tabouret G, Chabert A, PrPSc accumulation in myocytes from sheep incubating natural scrapie. Nat Med. 2004;10:591–3. DOIPubMedGoogle Scholar
- Espinosa JC, Andréoletti O, Castilla J, Herva ME, Morales M, Alamillo E, Sheep-passaged bovine spongiform encephalopathy agent exhibits altered pathobiological properties in bovine-PrP transgenic mice. J Virol. 2007;81:835–43. DOIPubMedGoogle Scholar
- Castilla J, Gutiérrez-Adán A, Brun A, Pintado B, Parra B, Ramirez MA, Different behavior toward bovine spongiform encephalopathy infection of bovine prion protein transgenic mice with one extra repeat octapeptide insert mutation. J Neurosci. 2004;24:2156–64. DOIPubMedGoogle Scholar
- Bruce ME, McConnell I, Fraser H, Dickinson AG. The disease characteristics of different strains of scrapie in Sinc congenic mouse lines: implications for the nature of the agent and host control of pathogenesis. J Gen Virol. 1991;72:595–603. DOIPubMedGoogle Scholar
- Bishop MT, Hart P, Aitchison L, Baybutt HN, Plinston C, Thomson V, Predicting susceptibility and incubation time of human-to-human transmission of vCJD. Lancet Neurol. 2006;5:393–8. DOIPubMedGoogle Scholar
- Padilla D, Béringue V, Espinosa JC, Andréoletti O, Jaumain E, Reine F, Sheep and goat BSE propagate more efficiently than cattle BSE in human PrP transgenic mice. PLoS Pathog. 2011;7:e1001319. DOIPubMedGoogle Scholar
Figures
Cite This ArticleTable of Contents – Volume 17, Number 9—September 2011
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Juan-María Torres, Centro de Investigación en Sanidad, Ctra. de Algete a El Casar, km. 8.100, 28130 Valdeolmos, Spain
Top