Volume 19, Number 4—April 2013
Letter
Novel Hantavirus in Wildlife, United Kingdom
Figure
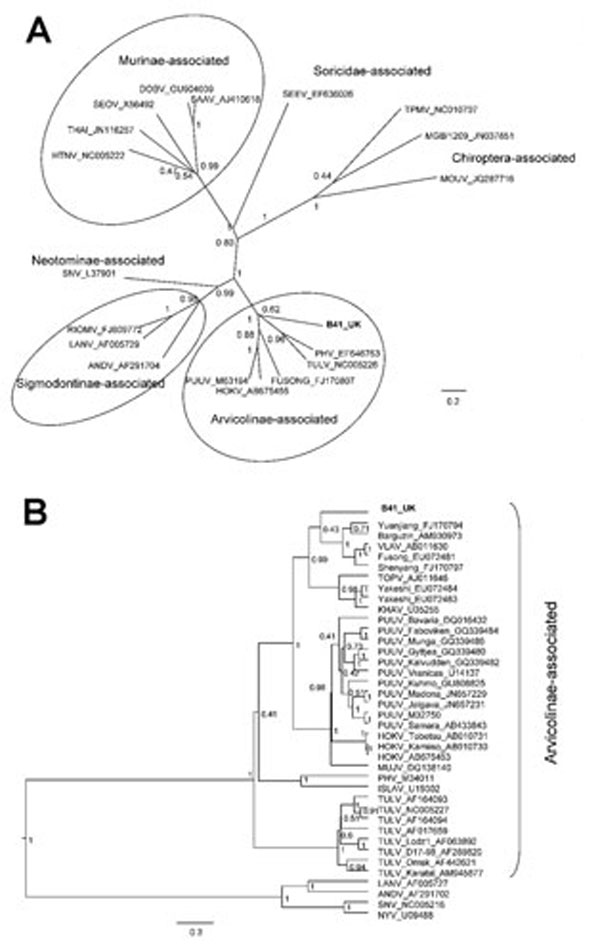
Figure. . . Bayesian phylogenetic trees constructed by using the models HKY+gamma for partial large segment sequences (n = 19) (A) and GTR+gamma for partial small segment sequences (n = 39) (B) within BEAST software (6) with Markov chain Monte Carlo chain lengths of 10 million and strict clock. Optimum substitution models were estimated by using MEGA5 (7). The trees are drawn to scale; branch lengths are measured in the number of substitutions per site. The numbers at each node are posterior probabilities. All effective sample size values exceeded 150 for partial L and 1,600 for partial S sequences. The phylogenetic position of virus isolated from field vole B41 (in boldface) is shown in relation to representative hantaviruses (A) and more closely related Arvicolinae-associated hantaviruses (B). GenBank accession numbers are shown next to taxonomic names. Scale bars indicate nucleotide substitutions per site. VLAV, Vladivostok virus; TOPV, Topografov virus; KHAV, Khabarovsk virus; PUUV, Puumala virus; HOKV, Hokkaido virus; MUJV, Muju virus; PHV, Prospect Hill virus; ISLAV, Isla Vista virus; TULV, Tula virus; LANV, Laguna Negra virus; ANDV, Andes virus; SNV, Sin Nombre virus; NYV, New York virus.
References
- Vaheri A, Henttonen H, Voutilainen L, Mustonen J, Sironen T, Vapalahti O. Hantavirus infections in Europe and their impact on public health. Rev Med Virol. 2013;23:35–49.
- Olsson GE, Leirs H, Henttonen H. Hantaviruses and their hosts in Europe: reservoirs here and there, but not everywhere? Vector Borne Zoonotic Dis. 2010;10:549–61 . DOIPubMedGoogle Scholar
- Fhogartaigh CN, Newsholme W, Kinirons M, Tong W. An emerging infectious cause of renal impairment in the UK. BMJ Case Rep. 2011. pii: bcr0620114326.
- Klempa B, Fichet-Calvet E, Lecompte E, Auste B, Aniskin V, Meisel H, Hantavirus in African wood mouse, Guinea. Emerg Infect Dis. 2006;12:838–40 . DOIPubMedGoogle Scholar
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214 . DOIPubMedGoogle Scholar
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis using maximum-likelihood, evolutionary distance, and maximum-parsimony methods. Mol Biol Evol. 2011;28:2731–9. DOIPubMedGoogle Scholar
- Vaheri A, Vapalahti O, Plyusnin A. How to diagnose hantavirus infections and detect them in rodents and insectivores. Rev Med Virol. 2008;18:277–88. DOIPubMedGoogle Scholar
- Schlegel M, Ali HS, Stieger N, Groschup MH, Wolf R, Ulrich RG. Molecular identification of small mammal species using novel cytochrome B gene–derived degenerated primers. Biochem Genet. 2012;50:440–7. DOIPubMedGoogle Scholar
- Schmidt-Chanasit J, Essbauer S, Petraityte R, Yoshimatsu K, Tackmann K, Conraths FJ, Extensive host sharing of central European Tula virus. J Virol. 2010;84:459–74 . DOIPubMedGoogle Scholar