Volume 20, Number 4—April 2014
Letter
St. Louis Encephalitis Virus Infection in Woman, Peru
Figure
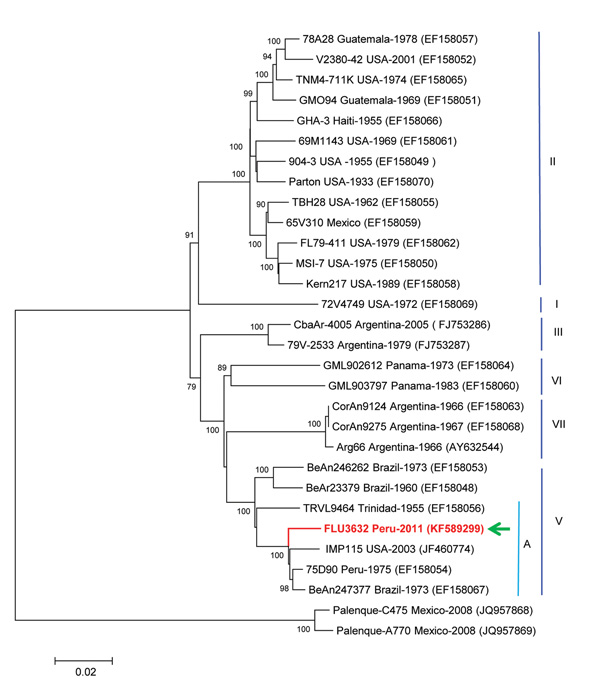
Figure. . Phylogenetic analysis from the initially sequenced 10,850-nt region of the St. Louis encephalitis virus (SLEV) genome, isolated from a woman in Peru, 2006. The sequence possessed only 92.8% homology with the NS5 gene region of the sole preexisting SLEV in the laboratory, a genotype II strain similar to TBH28 USA. The Peruvian SLEV sequence described in this case (FLU3632, arrow) groups with Brazil (1975), Peru (1973), and USA (2003) strains, inside the genotype V, subgenotype A. The evolutionary history was inferred by using the neighbor-joining method based on the Kimura 2-parameter model. The entire open reading frame sequence of the Peruvian SLEV isolate was determined in this study by using the Illumina HiSeq 1000 system (Illumina, Inc., San Diego, CA, USA) and assigned GenBank accession no. KF589299. Sequences were analyzed and assembled using the SeqMan Lasergene V.5 and then compared with sequences deposited in GenBank. Multiple sequence alignments were performed by using ClustalX version 2.0.10 (Conway Institute, University College, Dublin, Ireland; www.clustal.org/) and BioEdit version 7.0.9.0. (Ibis Biosciences, Carlsbad, CA, USA) Genetic divergence was determined by using MEGA version 5.02 (www.megasoftware.net/). Scale bar indicates nucleotide substitutions per site.