Volume 6, Number 5—October 2000
Research
Toxin Gene Expression by Shiga Toxin-Producing Escherichia coli: the Role of Antibiotics and the Bacterial SOS Response
Abstract
Toxin synthesis by Shiga toxin-producing Escherichia coli (STEC) appears to be coregulated through induction of the integrated bacteriophage that encodes the toxin gene. Phage production is linked to induction of the bacterial SOS response, a ubiquitous response to DNA damage. SOS-inducing antimicrobial agents, particularly the quinolones, trimethoprim, and furazolidone, were shown to induce toxin gene expression in studies of their effects on a reporter STEC strain carrying a chromosome-based stx2::lacZ transcriptional fusion. At antimicrobial levels above those required to inhibit bacterial replication, these agents are potent inducers (up to 140-fold) of the transcription of type 2 Shiga toxin genes (stx2); therefore, they should be avoided in treating patients with potential or confirmed STEC infections. Other agents (20 studied) and incubation conditions produced significant but less striking effects on stx2 transcription; positive and negative influences were observed. SOS-mediated induction of toxin synthesis also provides a mechanism that could exacerbate STEC infections and increase dissemination of stx genes. These features and the use of SOS-inducing antibiotics in clinical practice and animal husbandry may account for the recent emergence of STEC disease.
The associations between Escherichia coli O157:H7 infection, hemorrhagic colitis, and hemolytic uremic syndrome (HUS) were established in the early 1980s (1,2). Shiga toxin-producing E. coli (STEC) strains have since been recognized as the cause of both outbreaks and sporadic cases of diarrhea and HUS, involving thousands of cases and numerous deaths (3). Shiga toxins are key virulence factors in the pathogenesis of STEC disease (3). The term Shiga toxin (Stx) refers to two families of related toxins, Stx/Stx1, which includes the classical Shiga toxin produced by Shigella dysenteriae, and Stx2 (4). The stx genes carried by STEC strains are, with one possible exception (stx2e), encoded on bacteriophage genomes integrated into the bacterial chromosome. Stx2-producing STEC strains are more closely associated with HUS than are strains that produce only Stx1 (5,6).
Because antimicrobial agents may play a role in the pathogenesis of severe STEC disease, chemotherapy for STEC infections remains controversial (7,8). The location of stx genes (predominantly on λ-like bacteriophage genomes integrated into the chromosome of their host bacterium) has important implications because the induction of the SOS response, an extensively characterized genetic regulatory mechanism, induces high-level expression of previously silent bacteriophage genes (9). Stx genes are coexpressed with genes of the bacteriophage (10,11), and certain quinolones (known to be potent SOS inducers) induce increases in toxin (12,13) and bacteriophage production (13) of two to three orders of magnitude within 2 to 4 hours. The potential importance a link between the SOS response and prophage induction for Stx1 and Stx2 expression has been reinforced by recent sequencing and pathogenicity studies (11,14,15).
We have constructed a genetically modified derivative of a clinical isolate in which the genes encoding both elements of the toxin (stx2AB) were partially replaced with a lacZ reporter gene. The product of this gene, β-galactosidase, is easily assayed and detected; its expression reflects the transcriptional activity of the stx2 gene and can be visualized in simple agar plate assays and quantified in biochemical assays. We have extended our earlier observations on the effects of quinolones on reporter expression by this strain to include a wider range of antimicrobial agents and the modulating effects of different environmental conditions. Our results show that several agents could increase the amounts of toxin produced and that SOS-inducing agents could play an important role in the epidemiology of STEC infections.
Bacterial Strain and Growth Conditions
RV31, a strain of E. coli O157:H7 isolated locally from a patient with hemorrhagic diarrhea, was used to construct the reporter strain, PK552(stx2A::lacZ), which contains a copy of the E. coli lacZ gene transcriptionally fused to the promoter region of stx2 on the chromosome. The reporter strain construction, which involved allelic exchange with a series of suicide plasmid vectors, will be described elsewhere. In the first step, the entire indigenous lac operon and adjacent lacI gene were deleted to create a Lac- strain. Then, most of the coding sequence of the stx2A gene was replaced with a promoterless E. coli lacZ gene, resulting in a transcriptional fusion in which transcription of the lacZ reporter gene is controlled by stx2 regulatory mechanisms. The structure of the final construct was confirmed by diagnostic polymerase chain reaction, Southern blotting, and nucleotide sequencing across the lacZ insertion points.
Bacterial growth in the presence of antibiotics was monitored by the Bioscreen C system and associated Biolink software (Labsystems, Basingstoke, UK). This procedure allowed the simultaneous measurement of growth in up to 200 tests by recording changes in optical density. The stx2::lacZ reporter strain PK552 was grown overnight at 37°C in Antibiotic Medium No. 3 (Oxoid, Basingstoke, Hampshire, UK). A 2-mL aliquot of overnight culture was added to 100 mL of prewarmed medium and incubated with shaking at 37°C to an OD600nm of approximately 0.4. The culture was then divided, and test antibiotics were added to these aliquots from stock solutions. Aliquots of these cultures (200 µL, 4 replicates) were then injected into wells of honeycomb plates (Labsystems). The Biolink software was used to program the Bioscreen C system to specify wavelength, incubation temperature, length of experiment, timing of readings, and the rate of shaking of the cultures. The cultures were incubated in the Bioscreen C system at 37°C with continuous shaking, and OD600nm was recorded every 15 min. MICs for selected antibiotics were determined in the Bioscreen system by using the inoculation pattern described above and by the conventional agar dilution method.
Detection of β-galactosidase activity
An agar plate assay was used to screen the responses of the reporter strain, PK552 (stx2A::lacZ), in producing β-galactosidase activity in the presence of antibiotics. The assays were performed by using Luria-Bertani (LB) agar plates containing 20 g/mL of the chromogenic lactose analog X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside; Sigma, Poole, Dorset, UK). An agar overlay of the PK552 reporter strain was made by mixing 100 µL of an overnight culture with 3 mL of molten LB top agar, cooled to 48°C, and pouring the mixture over the surface of an LB agar plate. The plate was incubated with antibiotic discs (see below) placed on the surface, under the conditions specified. Expression of -galactosidase was indicated by the enzymatic cleavage of X-gal, resulting in a blue color, the intensity of which related to the amount of enzyme produced.
Microaerobic conditions (5% O2, 10% CO2, 85% N2) at 37°C were produced in a VAIN incubator (Don Whitley Scientific, Shipley, Yorkshire, UK). Microaerobic conditions at 42°C and anaerobic conditions were produced in gas jars by using Oxoid gas-generating kits according to the manufacturer's instructions.
Antibiotic discs (Oxoid/Unipath, UK) were as follows: ofloxacin (5 µg), nalidixic acid (30 µg), cinoxacin (100 µg), enrofloxacin (5 µg): flumequine (30 µg), ciprofloxacin (1 µg), perfloxacin (5 µg), norfloxacin (10 µg), amoxycillin/ clavulanic acid (20/10 µg), imipenem (10 µg), aztreonam (30 µg), ceftazidime (30 µg), cefotaxime (30 µg), cefuroxime (5 µg), piperacillin/tazobactam (10/75 µg), ampicillin (10 µg), cephalexin (30 µg), chloramphenicol (30 µg), doxycycline (30 µg), erythromycin (15 µg), trimethoprim (5 µg), sulphamethoxazole (25 µg), furazolidone (50 µg), amoxycillin (25 µg), novobiocin (30 µg), rifampin (25 µg), gentamicin (10 µg), fosfomycin (200 µg) (oral and systemic salts), polymyxin B (300 IU), and metronidazole (50 µg).
The Bioscreen C system was also used to measure total β-galactosidase activity in whole cultures of strain PK552 by orthonitrophenyl-β-D-galactoside (ONPG) assay (16). Aliquots (20 µL, 4 replicates) of culture were removed from wells of the honeycomb plate and added to a fresh plate. To this, 180 µL of Z buffer (60 mM Na2HPO4 7H2O, 40 mM NaH2PO4 H2O, 10 mM KCl, 1 mM MgSO4, and 50 mM β-mercaptoethanol, pH 7.0) containing 2 mg/mL lysozyme, 0.01% SDS, and 100 µg/mL chloramphenicol was added to the cells. The plate was incubated at 37°C for 30 min to lyse the cells and placed on ice until use. The Bioscreen C system was prewarmed to 28°C, and 40 µL of 4 mg/mL ONPG solution (made up in Z buffer) was added to each well. Plates were incubated in the Bioscreen C system at 28°C for 4 hours, and OD was recorded at both 420 nm and 540 nm every 10 min. β-galactosidase activity was determined by using the linear portion of the corrected OD420 /time relationship by the Miller formula, adjusted for the sample volume (16). Replicate samples (at least four in all assays reported) yielded mean values with coefficients of variation <10% in all cases.
Figure 1
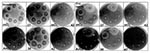
Figure 1. Zones of stx2 expression induced by various antimicrobial agents under different incubation conditions. The disc-diffusion assay plates demonstrate background levels and zones of blue coloration related to reporter gene β-galactosidase activity...
All the quinolones induced reporter expression, while only a few of the other agents had this effect (Figure 1). The induction occurred in three general patterns: a defined zone within the zone of growth inhibition (quinolones), a defined zone at the growth/no growth interface (furazolidone), or a diffuse zone within the zone of inhibition (trimethoprim). In addition, the incubation conditions appeared to produce a background level of induction or suppression. Microaerobic conditions and to a lesser extent incubation at 30°C were associated with background induction, while anaerobic conditions and incubation at 42°C had a suppressive effect as determined by the intensities of the blue zones. However, some quinolone-mediated induction was always detectable, even under the most suppressive condition (42°C). Induction by furazolidone and trimethoprim was, in general, similarly enhanced and suppressed, although 42°C abolished all induction and anaerobic conditions did not suppress trimethoprim/sulphamethoxazole induction.
As a group, only the agents that inhibit prokaryotic translation failed to induce reporter expression under any condition tested (Table). Like furazolidone, although less intensely, several β-lactam agents induced expression at the growth/no growth interface. Imipenem (a carbapenem) not only failed to induce expression but also inhibited induction by the adjacent monobactam, aztreonam (not shown), at subgrowth-inhibitory levels. We subsequently observed that this suppressive effect was confined to β-lactam-mediated induction (i.e., imipenem showed no suppressive effect against quinolones, trimethoprim, or furazolidone). We also noted an apparent inhibitory effect of clavulanic acid on amoxycillin in the co-amoxyclav combination and a requirement for potentiating conditions (30°C, mAO2 37°C) for detectable induction by novobiocin, polymixin B, and rifampin.
Figure 2

Figure 2. Dose-response characteristics of antibiotic-induced expression of stx2::lacZ. Exponential-phase inocula were exposed to antibiotics for 4 h (white bars) or 24 h (black bars), and β-galactosidase activity was determined at the end...
Figure 3

Figure 3. Time course of stx::lacZ induction by antibiotics. Exponential-phase cultures were exposed to the antibiotics indicated for up to 12 h, and whole culture β-galactosidase activities were determined. The exposure levels were...
We examined some of the features of induction by quantitative assay of β-galactosidase in cultures induced by exposure to antibiotics in liquid medium (Figure 2,3). The relationship of induction to MIC was consistent with that observed in the plate assays. Cefuroxime and furazolidone showed induction at sub-MIC levels, while the highest levels of induction with ofloxacin and trimethoprim were seen at supra-MIC exposure levels; the latter two agents were clearly more potent inducers (Figure 2,3). We also examined the effects of fosfomycin (0.03-80 g/L) in this assay; no inducing effect was detected.
The time course experiments (Figure 3) confirm the relative inducing potencies (Figure 2) and show that furazolidone-mediated induction is complete by 8 h. Levels of furazolidone-induced enzyme activity were lower at 24 h than at 4 h or 12 h. In contrast, trimethoprim and ofloxacin induction rapidly increased up to 6 h and 8 h, respectively, then continued to increase more slowly until the final measurement at 12 h. Additional expression induced by these two agents continued up to 24 h (Figure 2), while cefuroxime-induced expression occurred only between 12 h and 24 h.
These patterns of reporter gene expression demonstrate that several antimicrobial agents have the capacity to increase the amount of Stx2 synthesized by our STEC strain. Levels of β-galactosidase expression in the reporter strain are closely correlated with biologically active toxin produced by the parental wild type strain (12). The demonstrated link to de novo gene expression distinguishes our study from earlier work in which it could not be determined whether increased toxin levels reflected release of preformed toxin by cell lysis or increased synthesis (17–20). The inductions we observed show how certain agents and environmental conditions may increase the amount of toxin in infected persons and that, in the absence of definitive clinical data, they provide a rational basis for avoiding certain agents in treating patients who may have STEC infections.
Several of the agents tested here--the quinolones (21), trimethoprim (22), furazolidone, and metronidazole (23)--have a recognized capacity to induce bacterial SOS response, which is initiated when damaged bacterial DNA interacts with and activates the multifunctional RecA protein. Activated RecA, in turn, causes the degradation of two key repressor proteins, LexA and CI. The resulting de-repression of genes regulated by LexA leads to the temporary arrest of DNA synthesis and cell division and the activation of error-prone DNA repair. In strains carrying an integrated λ-like bacteriophage, cleavage of the CI phage repressor/activator protein leads to the induction of previously silent phage-encoded genes (in this case including stx2A and stx2B), followed by production of phage particles and host bacterial cell lysis (9).
The most potent SOS inducers, the quinolones and trimethoprim, produced the most prominent effects in both agar and liquid culture assays. Metronidazole did not induce reporter expression under the conditions tested; however, this agent would not normally be expected to affect E. coli. Another potent SOS inducer, mitomycin C, which is known to increase toxin levels in vitro (24,25), tests positive in agar and broth induction tests with our reporter strain (results not shown). The potential importance of the SOS response to pathogenesis in vivo has recently been underlined by a study in which STEC strains with mutations in recA, a critical gene for SOS induction, were rendered avirulent in a mouse LD50 test (15).
The importance of antibiotic-induced Stx expression would be reinforced if the implicated agents were in some way associated with occurrence or severity of STEC disease. The results of available studies have conflicted with regard to the influence of antibiotics. The age groups studied, the timing of antibiotic therapy, and the range of agents used complicate the analyses. Nonetheless, use of quinolones and trimethoprim/sulphamethoxazole (26,27) has been implicated as a risk factor for HUS. In a recent clinical study, children treated with cephalosporins or trimethoprim-sulphamethoxazole had, respectively, 13.4- and 17.7-fold increases in risk for HUS (28). Our observations suggest that the net effect of one of these agents on the exposure of an infected patient to toxin depends on the stage of the infection, the number of organisms present at the time antibiotics are administered, the immediate environmental conditions of those organisms, and the time-concentration profile and bactericidal effect of the drug. This complex interplay of factors could render an SOS-inducing antibiotic clinically beneficial (e.g., if the numbers of infecting organisms were insufficient to produce substantial amounts of toxin and they were all killed by the first dose), neutral, or disadvantageous in different situations. Moreover, exposure of patients to other potential SOS-inducing agents (cf 9,23.) could further complicate the relationship between antibiotic use and severe STEC disease. A strong association between mitomycin C administration and HUS was detected in a study of "cancer-associated" HUS (29).
Grif and colleagues reported on the effects of sub-MIC levels of 13 antibiotics on release of biologically active toxin (as distinct from toxin gene expression) by three STEC strains into culture supernatants after overnight incubation (20). These authors observed substantial interstrain differences in the responses, as well as increased levels of toxin after exposure to several agents that had not previously been associated with SOS-inducing activity. Grif et al. did not distinguish between enhanced release of pre-formed toxin and increased toxin production, and the most potent inducing effects we have observed were at suprainhibitory levels of exposure. Nonetheless, we cannot rule out interstrain differences in susceptibility to SOS activation and the possibility that other induction mechanisms may be involved. Our observations on SOS induction by several β-lactam agents and the inhibitory effects of imipenem appear pertinent. Suppression of toxin expression by imipenem supports previous observations on toxin release (30). Paired disc-diffusion assays involving imipenem and other recognized SOS-inducing antibiotics indicated that this effect was confined to induction by β-lactam agents (Kimmitt, Harwood, and Barer, unpub. data). This finding, combined with the different time course of induction with cefuroxime, reinforces the view that there may be induction pathways distinct from the SOS response. Although the nature of the putative alternate induction pathway(s) remains obscure, the clear evidence for β-lactam-mediated induction may contraindicate use of the implicated agents in treating patients with STEC infection.
Although any increase in exposure of STEC-infected persons to Shiga toxins seems undesirable, SOS-mediated induction seems particularly hazardous because of its potential rapid effects. We report kinetics (Figure 3) somewhat slower than those we observed previously in the shaken conical flask incubations (12). The rapid build-up in toxin levels associated with SOS induction could facilitate entry of toxin into the bloodstream and subsequent disseminated effects on the kidneys and other organs. SOS-mediated induction also leads to dissemination of the toxin-encoding bacteriophage.
Matsushiro et al. observed parallel increases in toxin and bacteriophage counts in response to norfloxacin (13), and we have made similar observations with ofloxacin and trimethoprim (Kimmit, Harwood and Barer, unpubl data). Moreover, sequencing studies clearly indicate that expression of stx2 and bacteriophage genes is coordinately regulated (11,14). Hemorrhagic colitis and HUS attributable to STEC were recognized after trimethoprim and the 4-quinolones were introduced into human and veterinary clinical practice, and the substantial recent increase in reports of STEC disease follows expanded use of fluoroquinolones.
Fluoroquinolones and trimethoprim have been recommended for prophylaxis and treatment of travelers' diarrhea (31), and the former are often used to treat severe bacterial enteric infections. Our findings indicate that this approach may be inappropriate if STEC infection is a possibility. Furthermore, reports that certain antimicrobial agents may ameliorate or reduce symptoms or the frequency of life-threatening complications in STEC infections provide an incentive to find a rational basis for selection (8,27). Our results suggest that agents with SOS-inducing activity, antimicrobial or otherwise, should be avoided (Table).
We conclude that SOS-mediated induction of Shiga toxins and toxin-encoding bacteriophages may contribute to the emerging epidemiologic pattern of STEC disease. Many other bacterial virulence determinants are encoded on lysogenic bacteriophage genomes (32), and the issues raised here may have public health and clinical implications beyond the understanding of STEC disease.
Dr. Kimmitt is trained in medical microbiology and molecular biology. He is currently studying the molecular biology of chlamydia infection at the London School of Hygiene and Tropical Medicine.
Acknowledgments
We thank D. Jenkins for providing strain RV31 from a patient with hemorrhagic diarrhea.
This work was funded by The Department of Health, London (DH 243).
References
- Karmali MA, Petric M, Lim C, Fleming PC, Steele BT. Escherichia coli cytotoxin, haemolytic-uraemic syndrome, and haemorrhagic colitis. Lancet. 1983;2:1299–300. DOIPubMedGoogle Scholar
- Riley LW, Remis RS, Helgerson SD, McGee HB, Wells JG, Davis BR, Hemorrhagic colitis associated with a rare Escherichia coli serotype. N Engl J Med. 1983;308:681–5. DOIPubMedGoogle Scholar
- Scotland SM, Willshaw GA, Smith HR, Rowe B. Properties of strains of Escherichia coli belonging to serogroup O157 with special reference to production of Vero cytotoxins VT1 and VT2. Epidemiol Infect. 1987;99:613–24. DOIPubMedGoogle Scholar
- Ostroff SM, Tarr PI, Neill MA, Lewis JH, Hargrett-Bean N, Kobayashi JM. Toxin genotypes and plasmid profiles as determinants of systemic sequelae in Escherichia coli O157:H7 infections. J Infect Dis. 1989;160:994–8.PubMedGoogle Scholar
- Carter AO, Borczyk AA, Carlson JA, Harvey B, Hockin JC, Karmali MA, A severe outbreak of Escherichia coli O157:H7-associated hemorrhagic colitis in a nursing home. N Engl J Med. 1987;317:1496–500. DOIPubMedGoogle Scholar
- Proulx F, Seidman E. Is antibiotic therapy of mice and humans useful in Escherichia coli O157:H7 enteritis? Eur J Clin Microbiol Infect Dis. 1999;18:533–4. DOIPubMedGoogle Scholar
- Walker GC. The SOS response of Escherichia coli. In: F.C. Neidhardt, editor. Escherichia coli and Salmonella. Washington: ASM Press; 1996. p. 1400-16.
- Muhldorfer I, Hacker J, Keusch GT, Acheson DW, Tschape H, Kane AV. Regulation of the Shiga-like toxin II operon in Escherichia coli. Infect Immun. 1996;64:495–502.PubMedGoogle Scholar
- Neely MN, Friedman DI. Functional and genetic analysis of regulatory regions of coliphage H-19B: location of shiga-like toxin and lysis genes suggest a role for phage functions in toxin release. Mol Microbiol. 1998;28:1255–67. DOIPubMedGoogle Scholar
- Kimmitt PT, Harwood CR, Barer MR. Induction of type 2 shiga toxin synthesis in Escherichia coli O157 by 4-quinolones. Lancet. 1999;353:1588–9. DOIPubMedGoogle Scholar
- Matsushiro A, Sato K, Miyamoto H, Yamamura T, Honda T. Induction of prophages of enterohemorrhagic Escherichia coli O157:H7 with norfloxacin. J Bacteriol. 1999;181:2257–60.PubMedGoogle Scholar
- Plunkett G, Rose DJ, Durfee TJ, Blattner FR. Sequence of Shiga toxin 2 phage 933W from Escherichia coli O157:H7: Shiga toxin as a phage late-gene product. J Bacteriol. 1999;181:1767–78.PubMedGoogle Scholar
- Fuchs S, Muhldorfer I, Donohue-Rolfe A, Kerenyic M, Emody L, Alexiev R, Influence of RecA on in vivo virulence and Shiga toxin 2 production in Escherichia coli pathogens. Microb Pathog. 1999;27:13–23. DOIPubMedGoogle Scholar
- Miller JH. Experiments in molecular genetics. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory; 1972.
- Karch H, Strockbine NA, O'Brien AD. Growth of Escherichia coli in the presence of trimethoprim-sulfamethoxazole facilitates detection of Shiga-like toxin producing strains by colony blot assay. FEMS Microbiol Lett. 1986;35:141–5. DOIGoogle Scholar
- Yoh M, Frimpong EK, Honda T. Effect of antimicrobial agents, especially fosfomycin, on the production and release of Vero toxin by enterohaemorrhagic Escherichia coli O157:H7. FEMS Immunol Med Microbiol. 1997;19:57–64. DOIPubMedGoogle Scholar
- Walterspiel JN, Ashkenazi S, Morrow AL, Cleary TG. Effect of subinhibitory concentrations of antibiotics on extracellular Shiga-like toxin-I. Infection. 1992;20:25–9. DOIPubMedGoogle Scholar
- Grif K, Dierich MP, Karch H, Allerberger F. Strain-specific differences in the amount of Shiga toxin released from enterohaemorrhagic Escherichia coli O157 following exposure to subinhibitory concentrations of antimicrobial agents. Eur J Clin Microbiol Infect Dis. 1998;17:761–6. DOIPubMedGoogle Scholar
- Drlica K, Zhao XL. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev. 1997;61:377–94.PubMedGoogle Scholar
- Lewin CS, Amyes SG. The role of the SOS response in bacteria exposed to zidovudine or trimethoprim. J Med Microbiol. 1991;34:329–32. DOIPubMedGoogle Scholar
- Quillardet P, Hofnung M. The SOS chromotest: a review. Mutat Res. 1993;297:235–79.PubMedGoogle Scholar
- Yee AJ, De Grandis S, Gyles CL. Mitomycin-induced synthesis of a Shiga-like toxin from enteropathogenic Escherichia coli H.I.8. Infect Immun. 1993;61:4510–3.PubMedGoogle Scholar
- al-Jumaili I, Burke DA, Scotland SM, al-Mardini H, Record CO. A method of enhancing verocytotoxin production by Escherichia coli. FEMS Microbiol Lett. 1992;72:121–5. DOIPubMedGoogle Scholar
- Pavia AT, Nichols CR, Green DP, Tauxe RV, Mottice S, Greene KD, Hemolytic-uremic syndrome during an outbreak of Escherichia coli O157:H7 infections in institutions for mentally retarded persons: clinical and epidemiologic observations. J Pediatr. 1990;116:544–51. DOIPubMedGoogle Scholar
- Dundas S, Todd WTA. Clinical presentation, complications and treatment of infection with verocytotoxin producing Escherichia coli. Challenges for the clinician. J Appl Microbiol. 2000;88:24S–30S.PubMedGoogle Scholar
- Wong CS, Jelacic S, Habeeb RL, Watkins SL, Tarr PI. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N Engl J Med. 2000;342:1930–6. DOIPubMedGoogle Scholar
- Lesesne JB, Rothschild N, Erickson B, Korec S, Sisk R, Keller J, Cancer-associated hemolytic -uremic syndrome: analysis of 85 cases from a national registry. J Clin Oncol. 1989;115:781–9.PubMedGoogle Scholar
- Takahashi K, Narita K, Kato Y, Sugiyama T, Koide N, Yoshida T, Low-level release of Shiga-like toxin (verocytotoxin) and endotoxin from enterohemorrhagic Escherichia coli treated with imipenem. Antimicrob Agents Chemother. 1997;41:2295–6.PubMedGoogle Scholar
- Centers for Disease Control and Prevention. Health Information for International Travel 1999-2000. Atlanta, GA;1999. p. 168.
- Cheetham BF, Katz ME. A role for bacteriophages in the evolution and transfer of bacterial virulence determinants. Mol Microbiol. 1995;18:201–8. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleTable of Contents – Volume 6, Number 5—October 2000
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
M.R. Barer, Department of Microbiology and Immunology, University of Newcastle, The Medical School, Framlington Place, Newcastle upon Tyne, NE2 4HH, United Kingdom; Fax: +44 191 222 7736
Top