Volume 7, Number 3—June 2001
Synopsis
Spoligotype Database of Mycobacterium tuberculosis: Biogeographic Distribution of Shared Types and Epidemiologic and Phylogenetic Perspectives
Figure 3
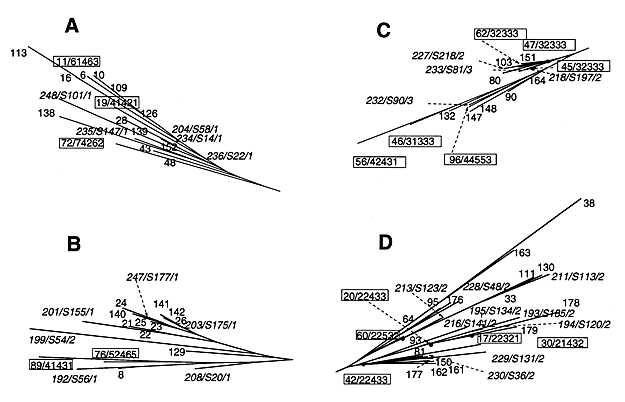
Figure 3. . Enlargement of branches A to D from the Mycobacterium tuberculosis phylogenetic tree (Figure 2). Numbers in standard characters refer to spoligotype numbers according to our database; those in boxes describe both the spoligotype number and variable number of tandem DNA repeats (VNTR) allele designations. Italicized numbers refer to spoligotype followed by the Houston spoligotype designation (12), and the major genetic groups 1 to 3 defined on the basis of katG283-gyrA95 allele combination (24). A and B show distinct branches belonging essentially to the major genetic group 1 with a high exact tandem repeat (ETR)-A copy number; C and D show branches that include some strains of the "Haarlem family" belonging to the major genetic group 2 with a low ETR-A copy number.
References
- World Health Organization. Global tuberculosis control. WHO report 1999. Geneva: The Organization; 1999.
- Slutkin G. Global AIDS 1981-1999: the response. Int J Tuberc Lung Dis. 2000;4:S24–33.PubMedGoogle Scholar
- Snider DE, Castro KG. The global threat of drug resistant tuberculosis. N Engl J Med. 1998;338:1689–90. DOIPubMedGoogle Scholar
- Long R, Nobert E, Chomyc S, van Embden J, McNamee C, Duran RR, Transcontinental spread of multidrug-resistant Mycobacterium bovis. Am J Respir Crit Care Med. 1999;159:2014–7.PubMedGoogle Scholar
- Bifani PJ, Mathema B, Liu Z, Moghazeh SL, Shopsin B, Tempalski B, Identification of a W variant outbreak of Mycobacterium tuberculosis via population-based molecular epidemiology. JAMA. 1999;282:2321–7. DOIPubMedGoogle Scholar
- Sola C, Devallois A, Horgen L, Maïsetti J, Filliol I, Legrand E, Tuberculosis in the Carribean: using spacer oligonucleotide typing to understand strain origin and transmission. Emerg Infect Dis. 1999;5:404–14. DOIPubMedGoogle Scholar
- Kamerbeek J, Schouls L, Kolk A, van Agterveld M, van Soolingen D, Kuijper S, Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol. 1997;35:907–14.PubMedGoogle Scholar
- Frothingham R, Meeker-O'Connell WA. Genetic diversity in the Mycobacterium tuberculosis complex based on variable numbers of tandem DNA repeats. Microbiology. 1998;144:1189–96. DOIPubMedGoogle Scholar
- Grimont PAD. TAXOTRON instruction manual. Paris: Taxolab, Institut Pasteur; 1996.
- Jaccard P. Nouvelles recherches sur la distribution florale. Bull Soc Vaud Sci Nat. 1908;44:223–70.
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–25.PubMedGoogle Scholar
- Soini H, Pan X, Amin A, Graviss EA, Siddiqui A, Musser JM. Characterization of Mycobacterium tuberculosis isolates from patients in Houston, Texas, by spoligotyping. J Clin Microbiol. 2000;38:669–76.PubMedGoogle Scholar
- Groenen PMA, Bunschoten AE, van Soolingen D, van Embden JDA. Nature of DNA polymorphism in the direct repeat cluster of Mycobacterium tuberculosis; application for strain differentiation by a novel typing method. Mol Microbiol. 1993;10:1057–65. DOIPubMedGoogle Scholar
- Hancock JM. The contribution of slippage-like processes to genome evolution. J Mol Evol. 1995;41:1038–47. DOIPubMedGoogle Scholar
- Kremer K, van Soolingen D, Frothingham R, Haas WH, Hermans PWM, Martin C, Comparison of methods based on different molecular epidemiologial markers for typing of Mycobacterium tuberculosis strains: interlaboratory study of discriminatory power and reproducibility. J Clin Microbiol. 1999;37:2607–18.PubMedGoogle Scholar
- Fang Z, Doig C, Kenna DT, Smittipat N, Palittapongarnpim P, Watt B, IS6110-mediated deletions of wild-type chromosomes of Mycobacterium tuberculosis. J Bacteriol. 1999;181:1014–20.PubMedGoogle Scholar
- Fang Z, Morrison N, Watt B, Doig C, Forbes KJ. IS6110 transposition and evolutionary scenario of the direct repeat locus in a group of closely related Mycobacterium tuberculosis strains. J Bacteriol. 1998;180:2102–9.PubMedGoogle Scholar
- Filliol I, Sola C, Rastogi N. Detection of a previously unamplified spacer within the DR locus of Mycobacterium tuberculosis: Epidemiological implications. J Clin Microbiol. 2000;38:1231–4.PubMedGoogle Scholar
- van Embden JDA, van Gorkom T, Kremer K, Jansen R, Van der Zeijst BAM, Schouls LM. Genetic variation and evolutionary origin of the direct repeat locus of Mycobacterium tuberculosis complex bacteria. J Bacteriol. 2000;182:2393–01. DOIPubMedGoogle Scholar
- Buikstra JE. Paleoepidemiology of tuberculosis in the Americas. In: Palfi G, Dutour O, Deak J, Hutas I, editors: Tuberculosis: past and present. Szeged, Hungary: Golden Book Publisher Ltd.; 1999. p. 479-94.
- Ortner DJ. Paleopathology: implications for the history and evolution of tuberculosis. In: Palfi G, Dutour O, Deak J, Hutas I, editors. Tuberculosis: past and present. Szeged, Hungary: Golden Book Publisher Ltd; 1999. p. 255-61.
- Kurepina NE, Sreevatsan S, Plikaytis BB, Bifani PB, Connell ND, Donneelly RJ, Characterization of the phylogenetic distribution and chromosomal insertion sites of five IS6110 elements in Mycobacterium tuberculosis: non random integration in the dnaA-dnaN region. Tuber Lung Dis. 1998;79:31–42. DOIPubMedGoogle Scholar
- Nei M. Phylogenetic analysis in molecular evolutionary genetics. Annu Rev Genet. 1996;30:371–403. DOIPubMedGoogle Scholar
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–25.PubMedGoogle Scholar
- Källenius G, Koivula T, Ghebremichael S, Hoffner SE, Norberg R, Svensson E, Evolution and clonal traits of Mycobacterium tuberculosis in Guinea-Bissau. J Clin Microbiol. 1999;37:3872–8.PubMedGoogle Scholar
- Sreevatsan S, Pan X, Stockbauer K, Connell N, Kreiswirth B, Whittam T, Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination. Proc Natl Acad Sci U S A. 1997;97:9869–74. DOIPubMedGoogle Scholar
- Castets M, Boisvert H, Grumbach F, Brunel M, Rist N. Les bacilles tuberculeux de type africain: note préliminaire. Rev Tuberc Pneumol (Paris). 1968;32:179–84.PubMedGoogle Scholar
- Frothingham R, Strickland PL, Bretzel G, Ramaswamy S, Musser JM, Williams DL. Phenotypic and genotypic characterization of Mycobacterium africanum isolates from West Africa. J Clin Microbiol. 1999;37:1921–6.PubMedGoogle Scholar
- Warren GM, Richardson M, Sampson S, Bourn W, van der Spuy G, Hide W, RFLP analysis of M. tuberculosis demonstrates strain-dependent evolution. Int J Tuberc Lung Dis. 1999;3(Suppl. I):S38.
- Tanaka MM, Small PM, Salamon H, Feldman MW. The dynamics of repeated elements: applications to the epidemiology of tuberculosis. Proc Natl Acad Sci U S A. 2000;97:3532–7. DOIPubMedGoogle Scholar
- Bauer J, Andersen AB, Kremer K, Miörner H. Usefulness of spoligotyping to discriminate IS6110 low-copy-number Mycobacterium tuberculosis complex strains cultures in Denmark. J Clin Microbiol. 1999;37:2602–6.PubMedGoogle Scholar
- Bonora S, Gutierrez MC, Perri GD, Brunello F, Allegranzi B, Ligozzi M, Comparative evaluation of Ligation-mediated PCR and spoligotyping as screening methods for genotyping of Mycobacterium tuberculosis strains. J Clin Microbiol. 1999;37:3118–23.PubMedGoogle Scholar
- Diaz R, Kremer K, de Haas PEW, Gomez RI, Marrero A, Valdivia JA, Molecular epidemiology of tuberculosis in Cuba outside of Havana, July 1994-June 1995: utility of spoligotyping versus IS6110 restriction fragment length polymorphism. Int J Tuberc Lung Dis. 1998;2:743–50.PubMedGoogle Scholar
- Douglas JT, Qian L, Montoya JC, Sreevatsan S, Musser J, van Soolingen D, Detection of a novel family of tuberculosis isolates in the Philippines. 97th general meeting of the American Society for Microbiology. Washington: ASM Press; 1997. p.572.
- Escalante P, Ramaswamy S, Sanabria H, Soini H, Pan X, Valiente-Castillo O, Genotypic characterization of drug-resistant Mycobacterium tuberculosis isolates from Peru. Tuber Lung Dis. 1998;79:111–8. DOIPubMedGoogle Scholar
- Goguet dela Salmonière YO, Li HM, Torrea G, Bunschoten A, van Embden JDA, Gicquel B. Evaluation of spoligotyping in a study of the transmission of Mycobacterium tuberculosis. J Clin Microbiol. 1997;35:2210–4.PubMedGoogle Scholar
- Goyal M, Saunders NA, van Embden JDA, Young DB, Shaw RJ. Differentiation of Mycobacterium tuberculosis isolates by spoligotyping and IS6110 restriction fragment length polymorphism. J Clin Microbiol. 1997;35:647–51.PubMedGoogle Scholar
- Heyderman RS, Goyal M, Roberts P, Ushewokunze S, Zishou S, Marshall BG, Pulmonary tuberculosis in Harare, Zimbabwe: analysis by spoligotyping. Thorax. 1998;53:346–50. DOIPubMedGoogle Scholar
- Niang MN, Goguet de la Salmonière YO, Samb A, Hane AA, Cisse MF, Gicquel B, Characterization of M. tuberculosis strains from West-African patients by spoligotyping. Microbes Infect. 1999;1:1189–92. DOIPubMedGoogle Scholar
- Popa MI, Goguet de la Salmonière YO, Teodor I, Popa L, Stefan M, Banica D, Genomic profile of Romanian M. tuberculosis strains appreciated by spoligotyping. Roum Arch Microbiol Immunol. 1997;56:63–75.PubMedGoogle Scholar
- de C. Ramos M. Soini H, Roscanni GC, Jaques M, Villares MC, Musser JM. Extensive cross-contamination of specimens with Mycobacterium tuberculosis in a reference laboratory. J Clin Microbiol. 1999;37:916–9.PubMedGoogle Scholar
- Taylor GM, Goyal M, Legge AJ, Shaw RJ, Young D. Genotypic analysis of Mycobacterium tuberculosis from medieval human remains. Microbiology. 1999;145:899–904. DOIPubMedGoogle Scholar
- van der Zanden AG, Hoentgen AH, Heilmann FG, Weltvreden EF, Schouls LM, van Embden JD. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis complex in paraffin wax embedded tissues and in stained microscopic preparations. Mol Pathol. 1998;51:209–14. DOIPubMedGoogle Scholar
- van Soolingen D, Qian L, de Haas PEW, Douglas JT, Traore H, Portaels F, Predominance of a single genotype of Mycobacterium tuberculosis in countries of East Asia. J Clin Microbiol. 1995;33:3234–8.PubMedGoogle Scholar
1For this purpose, the independent sampling sizes for Europe and the USA were taken as n1 and n2, the number of individuals within a given shared-type "x" was k1 and k2, and in this case, the representativeness of the two samples was p1=k1/n1 and P2=k2/n2, respectively. To assess if the divergence observed between p1 and p2 was due to sampling bias or the existence of two distinct populations, the percentage of individuals (p0) harboring shared-type "x" in the population studied was estimated by the equation p0= k1+k2/n1+n2=n1p1+n2p2/n1+n2. The distribution of the percentage of shared-type "x" in the sample sizes n1 and n2 follows a normal distribution with a mean p0 and a standard deviation of 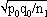 and
and 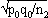 respectively, and the difference d=p1-p2 follows a normal distribution of mean p0-p0=0 and of variance σd2=σp12+σp22 = p0q0/n1+p0q0/n2 or σd2=p0q0 (1/n1+1/n2). The two samples being independent, the two variances were additive; the standard deviation σd=
respectively, and the difference d=p1-p2 follows a normal distribution of mean p0-p0=0 and of variance σd2=σp12+σp22 = p0q0/n1+p0q0/n2 or σd2=p0q0 (1/n1+1/n2). The two samples being independent, the two variances were additive; the standard deviation σd=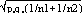 . If the absolute value of the quotient d/σd<2, the two samples were considered to belong to a same population (CI 95%) and the variation observed in the distribution of isolates for given shared types could be due to a sampling bias. Inversely, if d/σd>2, then the differences observed in the distribution of isolates for given shared types were statistically significant and not due to potential sample bias.
. If the absolute value of the quotient d/σd<2, the two samples were considered to belong to a same population (CI 95%) and the variation observed in the distribution of isolates for given shared types could be due to a sampling bias. Inversely, if d/σd>2, then the differences observed in the distribution of isolates for given shared types were statistically significant and not due to potential sample bias.