Volume 10, Number 8—August 2004
Research
Crimean-Congo Hemorrhagic Fever in Turkey
Figure 1
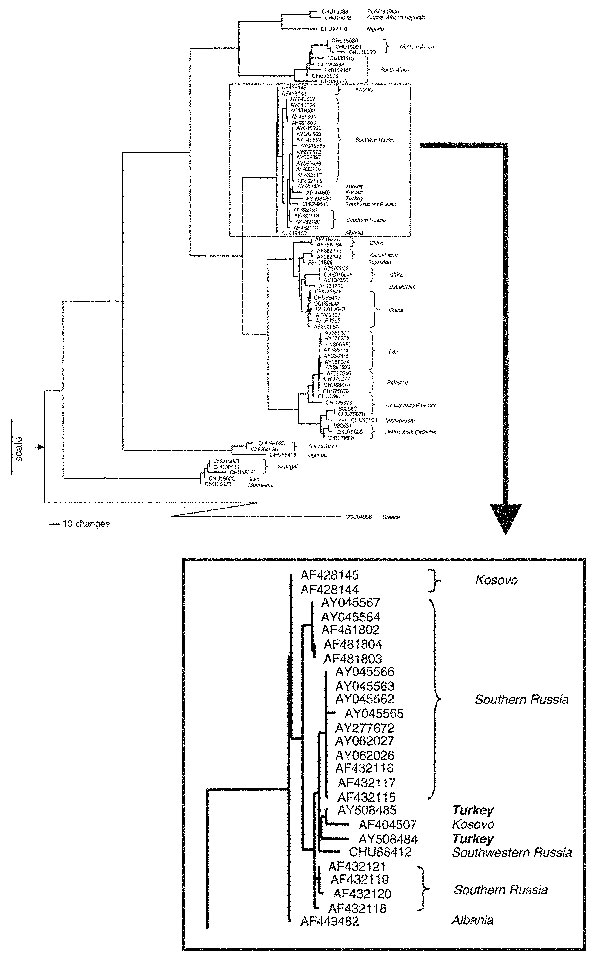
Figure 1. Phylogenetic analysis of Crimean-Congo hemorrhagic fever virus (CCHFV) genetic difference. Maximum parsimony analysis of the aligned sequences of a 488-nt region of CCHFV S segments and the equivalent genome region of Dugbe and Nairobi sheep disease viruses. Analysis was performed with the heuristic search method with stepwise addition, tree bisection-reconnection branch swapping, and transversions; transitions were weighted 4:1. The graphic representation of the results was outgroup rooted by using the Dugbe (GenBank accession no. AF434161, AF434162, AF434163, AF014014, AF434164, AF014015, AF434165) and Nairobi sheep disease virus (AF504293) S segment nucleotide sequences. The node attaching the outgroup to the CCHFV tree topology is shown by the arrow at the base of the tree. Horizontal distances within the CCHFV part of the tree are proportional to nucleotide steps (see scale bar), separating virus taxa and nodes. Vertical and diagonal lines are for visual clarity. Each virus sequence is indicated by the corresponding GenBank accession number. The two CCHFV sequences are in bold.