Volume 10, Number 8—August 2004
Dispatch
Crimean-Congo Hemorrhagic Fever in Bulgaria
Anna Papa* , Iva Christova†, Evangelia Papadimitriou*, and Antonis Antoniadis*
, Iva Christova†, Evangelia Papadimitriou*, and Antonis Antoniadis*
Figure 2
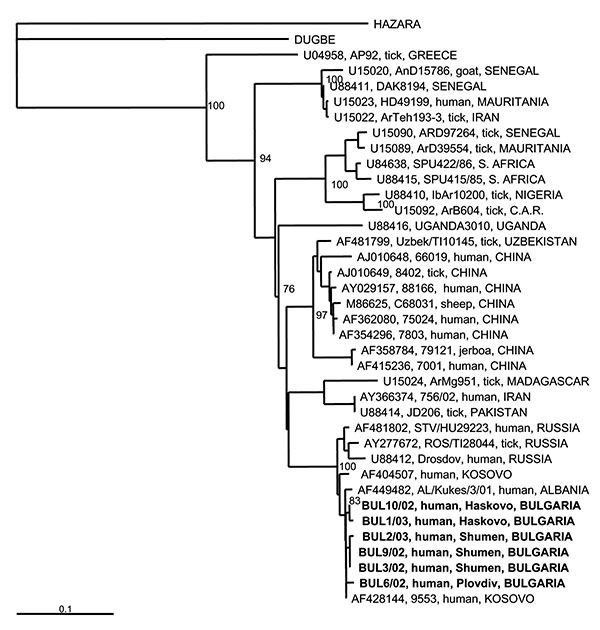
Figure 2. Phylogenetic tree based on 255-nt fragment from the S RNA segment, showing the clustering of the sequences obtained from this study and respective representative Crimean-Congo hemorrhagic fever virus strains from GenBank database. Sequences of two other nairoviruses, Dugbe and Hazara, were included; Hazara virus was used as outgroup. The numbers indicate percentage bootstrap replicates (of 100); values below 70% are not shown. Horizontal distances are proportional to the nucleotide differences. The scale bar indicates 10% nucleotide sequence divergence. Vertical distances are for clarity only. Sequences used in the analysis are indicated at the tree as: GenBank accession no., strain, host, country. CAR, Central African Republic.
Page created: March 03, 2011
Page updated: March 03, 2011
Page reviewed: March 03, 2011
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.