Volume 13, Number 4—April 2007
Dispatch
Genetic Stasis of Dominant West Nile Virus Genotype, Houston, Texas
Figure
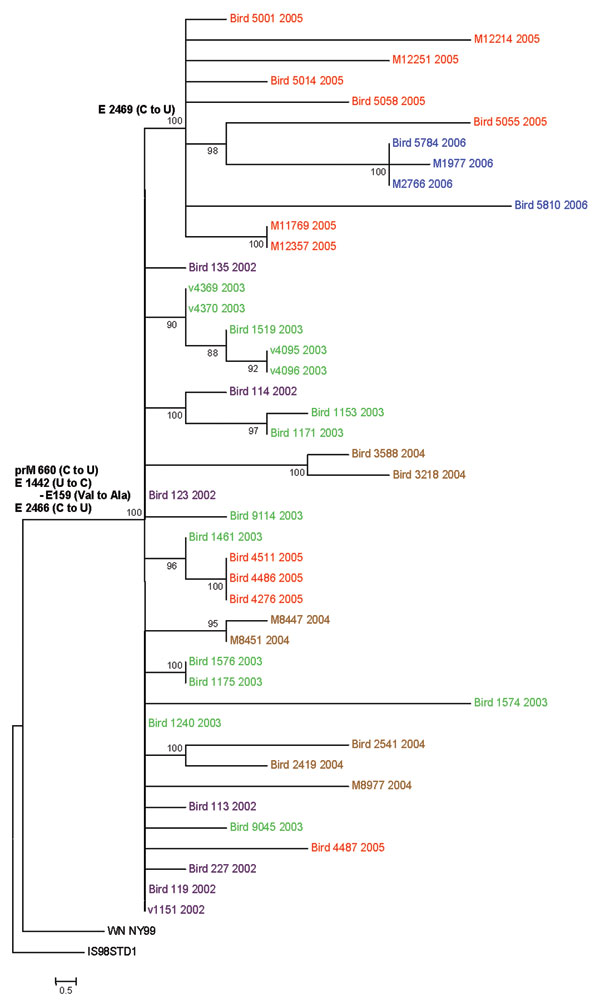
Figure. Phylogenetic tree generated by maximum likelihood analysis of a nucleotide alignment of the premembrane and E protein genes (2004 nucleotides) of previous and newly sequenced West Nile virus (WNV) isolates collected in the Houston metropolitan area from 2002 to 2006. The tree was rooted with the most closely related Old World WNV strain, Israel-1998. Maximum likelihood analysis was used to generate trees using PAUP (Version 4.0b11, Sinauer Associates, Sunderland, MA, USA) under the general time-reversible model and a γ distribution of substitution rates with statistical support and tree topology confirmation provided by 1,000 bootstrap replicates (bootstrap values shown at each node). Parsimony informative nucleotide mutations and deduced amino acid substitutions responsible for the observed clade topologies were added to the tree at relevant nodes. The year of isolation is color coded for each isolate on the tree (2002, purple; 2003, green; 2004, brown; 2005, red; 2006, blue), and the scale bar represents 0.5 nt changes.