Volume 13, Number 5—May 2007
Dispatch
Chikungunya Virus, Cameroon, 2006
Figure
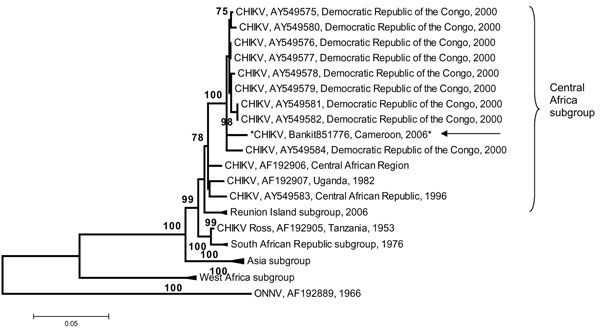
Figure. Phylogenetic tree of chikungunya virus (CHIKV) based on partial nucleotide sequences (3′ extremity of E1/3′-UTR, position 10,238–11,367). Phylogram was constructed with MEGA 2 program and tree drawing used the Jukes-Cantor algorithm for genetic distance determination and the neighbor-joining method. The percentage of successful bootstrap replicates (1,000 bootstrap replications, confidence probability >90%) is indicated at the nodes. The length of branches is proportional to the number of nucleotide changes (% of divergence). Asterisk (*) and arrow indicate the strains isolated in this work. The dark triangle corresponds to viruses clustering together. O’nyong-nyong virus (ONNV) sequence has been introduced for correct rooting of the tree. The GenBank reference no. for the Cameroon CHIKV isolate is EF051584.