Volume 13, Number 6—June 2007
Dispatch
Endemic Human Monkeypox, Democratic Republic of Congo, 2001–2004
Figure 2
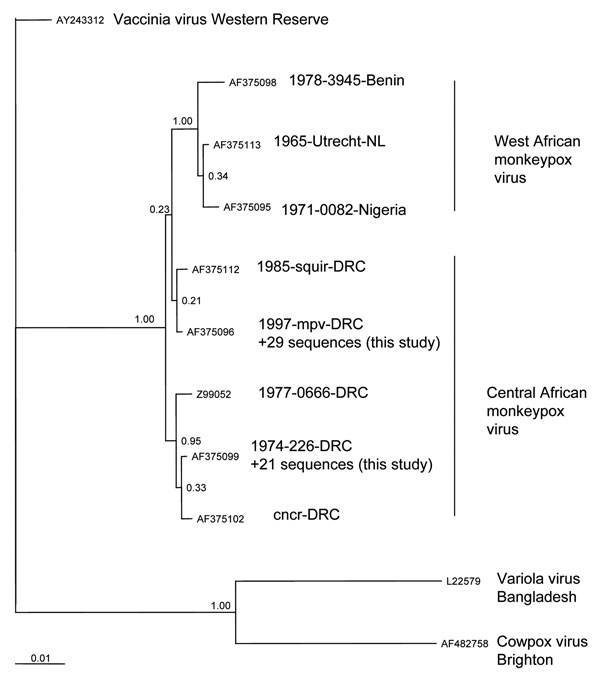
Figure 2. Phylogenetic inference relationships of the open reading frames encoding the hemagglutinin protein of selected strains of vaccinia, variola, cowpox, and monkeypox viruses and monkeypox virus isolates described in this study. Sequences used were cowpox virus Brighton (AF375089), variola virus Bangladesh (AF375129), vaccinia virus Lister (AY678276), monkeypox virus mpv 1997 (AF375096), mvp-squir (AF375112), mpv Zaire 77-0666 (Z99052), mpv-cncr (AF375102), mpv 74-226 (AF375099), mpv-082 (AF375095), mpv-utc (AF375113), and mpv-3945 (AF375098). ClustalW, version 1.83 (10), was used to generate amino acid multiple sequence alignments (pairwise gap opening = 35 and gap extension = 0.75; multiple alignment gap opening = 15 and gap extension = 0.30; Gonnet series). Each alignment was processed using RevTrans (11). Bayesian posterior probability inference of phylogeny used MrBayes, version 3.084. MrBayes settings for the best-fit model (GTR+I+G) were selected by hierarchies for the likelihood ratio test in MrModeltest 2.0 (12). Bayesian analysis was performed with MrBayes; the maximum likelihood model used 6 substitution types (nst = 6). Rate variation across sites was modeled by using a gamma distribution, with a proportion of sites being invariant (rates = invgamma). The Markov chain Monte Carlo search was run for 1 million generations; trees were sampled every 100 generations (the first 4,000 trees were discarded as burn-in). NL, the Netherlands; DRC, Democratic Republic of Congo.
References
- Jezek Z, Fenner F. Human Monkeypox. In: JL Melnick, editor. Monographs in virology. Volume 17. Basel: Karger; 1988.
- Centers for Disease Control and Prevention. Human monkeypox—Kasai Oriental, Democratic Republic of Congo, February 1996–October 1997. MMWR Morb Mortal Wkly Rep. 1997;46:1168–71.PubMedGoogle Scholar
- Hutin YJ, Williams RJ, Malfait P, Pebody R, Loparev VN, Ropp SL, Outbreak of human monkeypox, Democratic Republic of Congo, 1996 to 1997. Emerg Infect Dis. 2001;7:434–8.PubMedGoogle Scholar
- Di Giulio DB, Eckburg PB. Human monkeypox: an emerging zoonosis. Lancet Infect Dis. 2004;4:15–25. DOIPubMedGoogle Scholar
- Olson VA, Laue T, Laker MT, Babkin IV, Drosten C, Shelkunov SN, Real-time PCR system for detection of orthopoxviruses and simultaneous identification of smallpox virus. J Clin Microbiol. 2004;42:1940–6. DOIPubMedGoogle Scholar
- Meyer H, Perrichot M, Stemmler M, Emmerich P, Schmitz H, Varaine F, Outbreaks of disease suspected of being due to human monkeypox virus infection in the Democratic Republic of Congo in 2001. J Clin Microbiol. 2002;40:2919–21. DOIPubMedGoogle Scholar
- Hammarlund E, Lewis MW, Carter S, Amanna I, Hansen SG, Strelow LI, Multiple diagnostic techniques identify previously vaccinated individuals with protective immunity against monkeypox. Nat Med. 2005;11:1005–11.PubMedGoogle Scholar
- Damaso CR, Esposito JJ, Condit RC, Moussatche N. An emergent poxvirus from humans and cattle in Rio de Janeiro State: Cantagalo virus may derive from Brazilian smallpox vaccine. Virology. 2000;277:439–49. DOIPubMedGoogle Scholar
- Chen N, Li G, Liszewski MK, Atkinson JP, Jahrling PB, Feng Z, Virulence differences between monkeypox virus isolates from West Africa and the Congo basin. Virology. 2005;340:46–63. DOIPubMedGoogle Scholar
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL-X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–82. DOIPubMedGoogle Scholar
- Wernersson R, Pedersen AG. RevTrans: multiple alignment of coding DNA from aligned amino acid sequences. Nucleic Acids Res. 2003;31:3537–9. DOIPubMedGoogle Scholar
- Nylander JA, Ronquist F, Huelsenbeck JP, Nieves-Aldrey JL. Bayesian phylogenetic analysis of combined data. Syst Biol. 2004;53:47–67. DOIPubMedGoogle Scholar
- Reed KD, Melski JW, Graham MB, Regnery RL, Sotir MJ, Wegner MV, The detection of monkeypox in humans in the Western Hemisphere. N Engl J Med. 2005;350:342–50. DOIGoogle Scholar
- Learned LA, Reynolds MG, Wassa DW, Li Y, Olson VA, Karem K, Extended interhuman transmission of monkeypox in a hospital community in the Republic of the Congo, 2003. Am J Trop Med Hyg. 2005;73:428–34.PubMedGoogle Scholar
- Damon IK, Roth CE, Chowdhary V. Discovery of monkeypox in Sudan. N Engl J Med. 2006;355:962–3. DOIPubMedGoogle Scholar