Volume 15, Number 3—March 2009
Dispatch
Detection of Novel SARS-like and Other Coronaviruses in Bats from Kenya
Abstract
Diverse coronaviruses have been identified in bats from several continents but not from Africa. We identified group 1 and 2 coronaviruses in bats in Kenya, including SARS-related coronaviruses. The sequence diversity suggests that bats are well-established reservoirs for and likely sources of coronaviruses for many species, including humans.
The 2003 outbreak of severe acute respiratory syndrome (SARS) generated renewed interest in coronaviruses (CoV) and the source for the SARS CoV that caused the outbreak in humans (1). Serologic studies demonstrated that the virus had not previously circulated in human populations to any large extent and suggested a source of zoonotic origin (2–4). A likely natural viral reservoir for the virus was not identified until horseshoe bats (Rhinolophus spp.) in several regions in the People’s Republic of China were demonstrated to harbor SARS-like CoVs (5). Subsequently, a number of other SARS-like CoVs, as well as CoVs from antigenic groups I and II, were identified from bats in Asia, Europe, and North America, and coronavirus antibodies were detected in African bat species (6–11). It is not surprising that a growing number of CoVs have been detected in bats. To date, >60 viral species have been detected in bats because their biodiversity (second only to rodents), high population densities, wide distribution, and ability to fly over long distances allow them to harbor and easily spread multiple infectious agents. Bats have long been known as natural hosts for lyssaviruses and more recently have been recognized as potential reservoirs for emerging human pathogens, including Ebola, Marburg, Nipah, and Hendra viruses in addition to SARS-CoV (12,13).
Figure 1
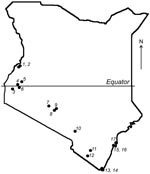
Figure 1. Map of Kenya showing the locations of 17 bat collection sites.
Given the association of bats with emerging infectious diseases, field surveys were performed during July–August 2006 in the southern portion of Kenya (Figure 1). The selection of sites was based on preliminary data regarding bat roost locations and observations of bats in the field during the survey. Attempts were made to collect specimens from 10–20 animals of each species present in each location. Bats were captured manually and by using mist nets and hand nets; adults and subadults of both sexes were captured. Each bat was measured, sexed, and identified to the genus or species level when possible. Blood samples and oral and fecal swabs were collected; the animals were then euthanized in compliance with field protocol. Blood, fecal swabs, and selected tissue samples were transported on dry ice from the field and stored at –80°C.
Fecal swabs (n = 221; Table) were screened for the presence of CoV RNA using 2 semi-nested reverse transcription–PCR (RT-PCR) assays. For the pan-coronavirus RT-PCR, conserved primers were designed from highly conserved regions of the RNA-dependent RNA polymerase (RdRp) gene 1b based on available CoV sequences (1st and 2nd round forward 5′-ATGGGITGGGAY TATCCWAARTGTG-3′; 1st round reverse 5′-AATTAT ARCAIACAACISYRTCRTCA-3′; 2nd round reverse 5′-CTAGTICCACCIGGYTTWANRTA-3′). For the pan–bat coronavirus RT-PCR, conserved primers were designed from the same highly conserved regions based on available bat CoV sequences and presumed to be more specific to bat coronaviruses (1st and 2nd round forward 5′-ATGGGITGGGAYTATCCWAARTGTG-3′; 1st round reverse 5′-TATTATARCAIACIACRCCATCRTC-3′; 2nd round reverse 5′-CTGGTICCACCI GGYTTNACRTA-3′). Total nucleic acids were extracted from 200 μL of a phosphate buffered saline suspension of each swab by using the QIAamp Mini Viral Elute kit (QIAGEN, Santa Clarita, CA, USA), according to the manufacturer’s instructions. The seminested RT-PCR was performed by using the SuperScript III One-Step RT-PCR kit and Platinum Tag Kit (Invitrogen, San Diego, CA, USA). The positive PCR products were purified by gel extraction by using the QIAquick Gel Extraction kit (QIAGEN) according to the manufacturer’s instructions; they were then sequenced on an ABI Prism 3130 automated sequencer (Applied Biosystems, Foster City, CA, USA), according to the manufacturer’s instructions.
Figure 2
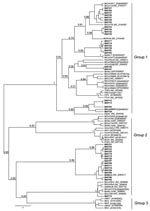
Figure 2. Phylogenetic tree generated using Bayesian Markov Chain Monte Carlo analysis implemented in Bayesian Evolutionary Analysis Sampling Trees (BEAST; http://beast.bio.ed.ac.uk) by using a 121-nt fragment of the RdRp gene 1b from...
Of 221 bat fecal swabs examined, 41 (19%) were positive by at least 1 of the 2 seminested RT-PCR assays (Table). One specimen had 2 distinct CoV sequences, each amplified by 1 of the 2 PCR assays, giving a total of 42 distinct CoV sequences. To characterize the overall diversity of CoV sequences, in this study a phylogenetic tree (Figure 2) of the 121-bp fragment of RdRp was generated from 39 coronaviruses from bats in Kenya and 47 selected human and animal coronaviruses from the National Center for Biotechnology Information database based on the Bayesian Monte Carlo Markov Chain method (14). Three of the 42 sequences were not of sufficiently high quality to include in this tree. Some nodes had low Bayesian posterior probabilities (Figure 2). Longer sequences from these viruses are needed to refine their phylogenetic relationships.
Among the 39 sequences in the tree, 23 belonged to previously defined group 1 and were mapped into 5 different sequence clusters. The 121-bp sequences in these 5 clusters had an average nucleic acid (NA) sequence identity of 88%, 85%, 81%, 77%, and 80% when compared with the next closest previously characterized CoVs (i.e., BtCoV1A, BtHKU8, BtHKU7, HCoV229E, and BtCoVA970, respectively). The remaining 16 sequences would likely be placed into group 2. Two sequences from Chaerophon spp. bats (location 17) were closely related to a SARS-like CoV cluster, including 1 sequence shown in Figure 2 (BtKY15) and another (BtKY16) that was 1 of the 3 low-quality sequences excluded from the tree. These 2 NA sequences show ≈89% identity with the nearest previously characterized bat: SARS-like CoV, BtCoVRF1, shows ≈80% NA sequence identity to SARS CoV (Urbani strain) and ≈63% NA sequence identity to the human group 2 CoV HCoVOC43. The 15 remaining NA sequences were grouped into 2 clusters. One cluster contains the recently described BtHKU9 with >95% NA sequence identity, and the other cluster (BtKY18-like cluster) contains no other previously known CoVs, with <75% NA sequence identity to BtHKU9.
The pattern of CoV detections by bat species and location demonstrates several features concerning coronaviruses in bats. A given bat species in the same location can harbor several distinct CoVs as noted for Chaerophon spp. (location 17), Miniopterus inflatus (location 5), and Rousettus aegyptiacus (location 2 and 16); similar CoVs can also been seen in the same type of bat in different locations, as noted for BtCoV1A-like cluster CoVs being detected in Miniopterus spp. bats of 4 species from different locations. One M. inflatus bat from location 5 harbored 2 different, but closely related, CoVs, 1 (BtCoV 36) from the BtCoV1A-like cluster and 1 (BtCoV 35) from the BtHKU8-like cluster (Figure 2). CoVs of these 2 closely related clusters were detected in Miniopterus spp. bats, but not detected in other bat genera, including those that shared roosts with Miniopterus spp. bats. This finding is consistent with studies from China in which BtCoV1A-like and BtHKU8-like CoVs were frequently identified but only in Miniopterus spp. bats (15). This may suggest that viruses of the BtCoV1A-like cluster and the BtHKU8-like cluster are specifically adapted to Miniopterus spp. bats and not easily transmitted to other bat species.
In contrast, other genetically similar CoVs were detected in several different bat species. For example, CoVs from th BtHKU7-like cluster were detected in both Chaerophon spp. and Otomops martinsseni bats; CoVs from the BtCoVA970-like cluster were detected in Cardioderma cor and Rousettus aegyptiacus bats; CoVs from the BtKY18-like cluster were detected in Chaerophon spp., Eidolon helvum, and R. aegyptiacus bats; and CoVs from the BtHKU9-like cluster were detected in Hipposidereos commersoni and R. aegyptiacus bats.
These data demonstrate that the CoV diversity in bats previously detected in Asia, Europe, and North America is also present, possibly to a greater extent, in Africa. The extent of this diversity among CoVs may be shown more clearly through additional studies in bats, and increased demonstration of CoV diversity in bats may require a reconsideration of how they should be grouped. The frequency and diversity of CoV detections in bats, now in multiple continents, demonstrate that bats are likely an important source for introduction into other species globally. Understanding the extent and diversity of CoV infection in bats provides a foundation for detecting new disease introductions that may, like SARS, present a public health threat.
Dr Tong is a virologist in the Gastroenteritis and Respiratory Virus Laboratory Branch of the Division of Viral Diseases, Centers for Disease Control and Prevention. Her interests are in novel emerging CoVs.
Acknowledgments
We thank Janet C. Beagley, Evelyne Mulama, Heather Burke, Dorine Bonyo, Edwin Danga, Leonard Nderitu, Solomon Gikundi, Lydia Kigo, M. Kariuki Njenga, and Olga Urazova for excellent technical and logistical assistance. We thank Yu Li, Jane Iber, and Cara Burns for their discussion and support in the phylogenetic analysis.
The study was supported in part by the Global Disease Detection program (Centers for Disease Control and Prevention, Atlanta, Georgia).
References
- Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348:1953–66. DOIPubMedGoogle Scholar
- Che XY, Hao W, Qiu LW, Pan YX, Liao ZY, Xu H, Antibody response of patients with severe acute respiratory syndrome (SARS) to nucleocapsid antigen of SARS-associated coronavirus [in Chinese]. Academic Journal of the First Medical College of PLA. 2003;23:637–9.PubMedGoogle Scholar
- Woo PC, Lau SK, Yuen KY. Infectious diseases emerging from Chinese wet-markets: zoonotic origins of severe respiratory viral infections. Curr Opin Infect Dis. 2006;19:401–7. DOIPubMedGoogle Scholar
- Guan Y, Zheng BJ, He YQ, Liu XL, Zhuang ZX, Cheung CL, Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003;302:276–8. DOIPubMedGoogle Scholar
- Lau SK, Woo PC, Li KS, Huang Y, Tsoi HW, Wong BH, Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc Natl Acad Sci U S A. 2005;102:14040–5. DOIPubMedGoogle Scholar
- Poon LL, Chu DK, Chan KH, Wong OK, Ellis TM, Leung YH, Identification of a novel coronavirus in bats. J Virol. 2005;79:2001–9. DOIPubMedGoogle Scholar
- 7. Müller MA, Paweska JT, Leman PA, Drosten C, Grywna K, Kemp A, et al. Coronavirus antibodies in African bat species. Emerg Infect Dis. 2007;13:1367–70.
- Gloza-Rausch F, Ipsen A, Seebens A, Gottsche M, Panning M, Felix Drexler J, Detection and prevalence patterns of group I coronaviruses in bats, northern Germany. Emerg Infect Dis. 2008;14:626–31. DOIPubMedGoogle Scholar
- Dominguez SR, O’Shea TJ, Oko LM, Holmes KV. Detection of group 1 coronaviruses in bats in North America. Emerg Infect Dis. 2007;13:1295–300.PubMedGoogle Scholar
- Tang XC, Zhang JX, Zhang SY, Wang P, Fan XH, Li LF, Prevalence and genetic diversity of coronaviruses in bats from China. J Virol. 2006;80:7481–90. DOIPubMedGoogle Scholar
- Woo PC, Lau SK, Li KS, Poon RW, Wong BH, Tsoi HW, Molecular diversity of coronaviruses in bats. Virology. 2006;351:180–7. DOIPubMedGoogle Scholar
- Wong S, Lau S, Woo P, Yuen KY. Bats as a continuing source of emerging infections in humans. Rev Med Virol. 2007;17:67–91. DOIPubMedGoogle Scholar
- Leroy EM, Kumulungui B, Pourrut X, Rouquet P, Hassanin A, Yaba P, Fruit bats as reservoirs of Ebola virus. Nature. 2005;438:575–6. DOIPubMedGoogle Scholar
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. DOIPubMedGoogle Scholar
- Chu DK, Poon LL, Chan KH, Chen H, Guan Y, Yuen KY, Coronaviruses in bent-winged bats (Miniopterus spp.). J Gen Virol. 2006;87:2461–6. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleTable of Contents – Volume 15, Number 3—March 2009
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Suxiang Tong, Centers for Disease Control and Prevention, 1600 Clifton Rd NE, Mailstop G18, Atlanta, GA 30333, USA;
Top