Volume 15, Number 9—September 2009
Letter
Genomic Diversity of Oseltamivir-Resistant Influenza Virus A (H1N1), Luxembourg, 2007–08
Figure
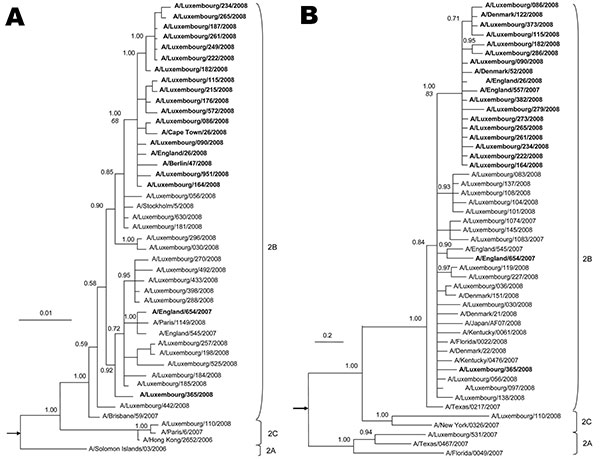
Figure. Phylogeny of A) neuraminidase (NA, complete gene) and B) polymerase complex 2 (C-terminal 1,300 nt) genes for selected influenza viruses A (H1N1) from Luxembourg and other countries. Subclades are identified to the right of each tree. The best-approximating model of nucleotide evolution was the general time reversible model with a gamma rate distribution and this model was used for the Bayesian analysis. Markov chain Monte Carlo sampling was implemented in MrBayes version 3 (8). In all cases, 6 chains with at least 4 million generations were calculated (10% burn-in removed). At least 2 independent runs of each analysis were performed. Posterior probabilities (indicated on important nodes) of the consensus tree topologies were estimated by sampling likelihood parameters every 125 generations. Boldface indicates sequences of oseltamivir-resistant influenza viruses A (H1N1) with the Tyr275 mutation in NA. In MEGA version 4, a neighbor-joining tree with 10,000 replicates was generated to calculate bootstrap values, shown in italics on the node dividing resistant and sensitive strains. Scale bars indicate nucleotide substitutions per site. The trees are rooted on A/New Caledonia/01/1999 and A/BrevigMission/1918 (indicated by arrows).
References
- Recommended composition of influenza virus vaccines for use in the 2009 southern hemisphere influenza season. Wkly Epidemiol Rec. 2008;83:366–72.PubMedGoogle Scholar
- World Health Organization. Influenza A(H1N1) virus resistance to oseltamivir—2008/2009 influenza season, northern hemisphere [cited 2009 Mar 25]. Available from http://www.who.int/csr/disease/influenza/H1N1webupdate20090318 ed_ns.pdf
- Meijer A, Lackenby A, Hungnes O, Lina B, van der Werf S, Schweiger B, Oseltamivir-resistant influenza A (H1N1) virus, Europe, 2007–08 season. Emerg Infect Dis. 2009;15:552–60. DOIPubMedGoogle Scholar
- Rameix-Welti MA, Enouf V, Cuvelier F, Jeannin P, van der Werf S. Enzymatic properties of the neuraminidase of seasonal H1N1 influenza viruses provide insights for the emergence of natural resistance to oseltamivir. PLoS Pathog. 2008;4:e1000103. DOIPubMedGoogle Scholar
- McKimm-Breschkin J, Trivedi T, Hampson A, Hay A, Klimov A, Tashiro M, Neuraminidase sequence analysis and susceptibilities of influenza virus clinical isolates to zanamivir and oseltamivir. Antimicrob Agents Chemother. 2003;47:2264–72. DOIPubMedGoogle Scholar
- Yen HL, Herlocher LM, Hoffmann E, Matrosovich MN, Monto AS, Webster RG, Neuraminidase inhibitor-resistant influenza viruses may differ substantially in fitness and transmissibility. Antimicrob Agents Chemother. 2005;49:4075–84. DOIPubMedGoogle Scholar
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–9. DOIPubMedGoogle Scholar
- Ronquist F, Huelsenbeck JP. MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–4. DOIPubMedGoogle Scholar
- Guilligay D, Tarendeau F, Resa-Infante P, Coloma R, Crepin T, Sehr P, The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat Struct Mol Biol. 2008;15:500–6. DOIPubMedGoogle Scholar
- Niman H. Emergence and fixing of antiviral resistance in influenza A via recombination and hitch hiking. 2009 Feb 10 [cited 2009 Mar 25]. Available from http://hdl.handle.net/10101/npre.2009.2832.1