Volume 17, Number 9—September 2011
CME ACTIVITY - Research
Mycobacterium chelonae-abscessus Complex Associated with Sinopulmonary Disease, Northeastern USA
Cite This Article
Citation for Media
Introduction
![]()
Medscape, LLC is pleased to provide online continuing medical education (CME) for this journal article, allowing clinicians the opportunity to earn CME credit.
This activity has been planned and implemented in accordance with the Essential Areas and policies of the Accreditation Council for Continuing Medical Education through the joint sponsorship of Medscape, LLC and Emerging Infectious Diseases. Medscape, LLC is accredited by the ACCME to provide continuing medical education for physicians.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1 AMA PRA Category 1 Credit(s)TM. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 70% minimum passing score and complete the evaluation at www.medscape.org/journal/eid; (4) view/print certificate.
Release date: August 22, 2011; Expiration date: August 22, 2012
Learning Objectives
Upon completion of this activity, participants will be able to:
• Analyze the M. chelonae-abscessus complex
• Distinguish the molecular identity of “M. franklinii”
• Identify the most common clinical source of “M. franklinii”
• Evaluate the antimicrobial susceptibility of “M. franklinii”
MEDSCAPE CME EDITOR
Beverly Merritt, Technical Writer/Editor, Emerging Infectious Diseases. Disclosure: Beverly Merritt has disclosed no relevant financial relationships.
MEDSCAPE CME AUTHOR
Charles P. Vega, MD, Associate Professor; Residency Director, Department of Family Medicine, University of California, Irvine. Disclosure: Charles P. Vega, MD, has disclosed no relevant financial relationships.
AUTHORS
Disclosures: Barbara A. Brown-Elliott, MS; Perry G. Ridge; Jacob D. Durtschi, BS; Linda Bridge Mann, BS; E. Susan Slechta; Arnold G. Steigerwalt, BS; Benjamin D. Moser; Anne M. Whitney, PhD; June M. Brown, BS; Karl V. Voelkerding, MD; Karin L. McGowan, PhD; Anne F. Reilly, MD, MPH; W. Ray Butler, MS; Paul H. Edelstein, MD; and Richard J. Wallace Jr., MD, have disclosed no relevant financial relationships. Keith E. Simmon, BS, has disclosed the following relevant financial relationships: employed by: Isentio US/Software company (non-FDA). Thomas J. Kirn, MD, has disclosed the following relevant financial relationships: served as an advisor or consultant for Merck Sharpe and Dohme. Cathy A. Petti, MD, has disclosed the following relevant financial relationships: was an employee for Novartis Diagnostics from 2009–2010.
Abstract
Members of the Mycobacterium chelonae-abscessus complex represent Mycobacterium species that cause invasive infections in immunocompetent and immunocompromised hosts. We report the detection of a new pathogen that had been misidentified as M. chelonae with an atypical antimicrobial drug susceptibility profile. The discovery prompted a multicenter investigation of 26 patients. Almost all patients were from the northeastern United States, and most had underlying sinus or pulmonary disease. Infected patients had clinical features similar to those with M. abscessus infections. Taxonomically, the new pathogen shared molecular identity with members of the M. chelonae-abscessus complex. Multilocus DNA target sequencing, DNA-DNA hybridization, and deep multilocus sequencing (43 full-length genes) support a new taxon for these microorganisms. Because most isolates originated in Pennsylvania, we propose the name M. franklinii sp. nov. This investigation underscores the need for accurate identification of Mycobacterium spp. to detect new pathogens implicated in human disease.
Infections caused by members of the Mycobacterium chelonae-abscessus complex remain a serious public health problem, and their role has expanded, with growing numbers of therapeutic interventions that disrupt the competency of the human immune system. Before 2001, only 2 species, M. chelonae and M. abscessus, were recognized as members of the complex. Since that time, 4 new species have been added to the complex: M. immunogenum, M. massiliense, M. bolletii, and M. salmoniphilum. With the exception of M. salmoniphilum, all members of the complex have been implicated in human disease (1–4). Recently investigators have noted a lack of separation of M. bolletii and M. massiliense from M. abscessus and have proposed they be classified as a subspecies of M. abscessus (5).
Members of the M. chelonae-abscessus complex represent Mycobacterium species that cause invasive skin and soft tissue infections, pneumonia, bloodstream infections, and abscesses in immunocompetent and immunocompromised hosts (6,7). Definitive identification by the clinical laboratory is needed for outbreak detection and for performance of susceptibility testing for patient management. Currently, the taxonomic relationships among members of the M. chelonae-abscessus complex lack clarity. The species are biochemically inert and their genetic signatures by partial 16S rRNA gene sequencing are often similar, which makes identification a great challenge for clinical laboratories.
In 2007, we detected a group of clinical isolates that were misidentified as M. chelonae with an atypical antimicrobial drug susceptibility profile. All isolates were from Pennsylvania, and as an interim identification, we labeled these isolates as CV for M. chelonae variant. Our discovery prompted a large multistate investigation that involved obtaining clinical correlation, retrospective and prospective collections of isolates with similar CV characteristics, and examination of a large set of known clinical isolates and type strains from the M. chelonae-abscessus complex. Given the taxonomic complexities and current ambiguities within the M. chelonae-abscessus group, we performed a comprehensive analysis of the potentially new species, including DNA-DNA hybridization, multilocus sequencing, and deep multilocus sequencing. We describe a new pathogen, M. franklinii sp. nov., a proposed new member of the M. chelonae-abscessus complex that was isolated from 26 patients in the United States. We discuss its potential role in human disease.
Isolates
All available type strains of M. chelonae-abscessus complex were obtained from American Type Culture Collection (ATCC), Collection of Institut Pasteur, or Culture Collection, University of Göteborg, Sweden. Previously identified clinical isolates of M. abscessus, and M. chelonae by partial 16S rRNA gene sequencing and internal transcribed spacer (ITS) PCR were retrieved for comparative analysis. A subset of these isolates was described in a prior study (8). CVs were defined as isolates that were cefoxitin susceptible or showed intermediate resistance, and identified as M. chelonae by partial 16S rRNA gene sequencing and ITS PCR (9) by Associated Regional and University Pathologists Laboratories and the Hospital of the University of Pennsylvania or by failure to amplify hsp65 by PCR restriction fragment length polymorphism analysis (10,11) by the University of Texas Health Science Center. Microbiologic and medical records were reviewed for clinical information for select isolates. Clinical case reviews were conducted under institutional review board approved protocols at Associated Regional and University Pathologists Laboratories, Children’s Hospital of Philadelphia, University of Pennsylvania, and the University of Texas Health Science Center. Study isolates CV002 (ATCC [pending] and DSMZ 45524) and CV005 (ATCC [pending]) have been deposited into culture collections.
Multilocus Sequencing
DNA extractions, PCR, and sequencing reactions were performed as described (8). Amplifications and sequencing reactions were performed by using primers specific for ≈1,400 bp of the 16S rRNA 5F (5′-TTGGAGAGTTTGATCCTGGCTC-3′) and 1492R (5′-ACGGITACCTTGTTACGACTT-3′), ≈700 bp of rpoB, ≈400 bp of sodA (12), ≈240 bp of the ITS (13), and ≈400 bp of hsp65 (11) genes or region. Sequence alignments and phylogenetic trees were constructed by using neighbor-joining method with Kimura 2-parameter distance correction model and 1,000 bootstrap replications in MEGA4 (14). Only unique sequences (sequevars) were included in the trees.
Standard for Identification
Final species identifications were based on comparisons of sequences for the full 16S rRNA and the partial rpoB genes to GenBank references of type strains. Full-length 16S rRNA sequences were used in this study, and we used 99.5% shared identity for identification to a type strain sequence. Species identification for rpoB gene was based on an identity of 98.0%–100% as outlined by Adekambi et al. (12,15).
DNA-DNA Hybridization
Purified DNA of the type strains and the patient isolates CV002, CV004, CV005, CV006, and CV005 was prepared as described (16). CV002 and CV005 strains were labeled with [32P] dCTP using the Nick Translation Kit (Invitrogen, Carlsbad, CA, USA). Labeled DNA from the CV002 was hybridized with unlabeled DNA from isolates CV002, CV004, CV005, CV006, and CV015 and with unlabeled DNA from the type strains. Labeled DNA from patient isolate CV005 was then hybridized with unlabeled DNA from isolates CV002 and CV005. The reciprocal experiment was performed because of the nearness of CV005 to the 70% cut-off designated by Wayne (17) and the 0% divergence obtained in the first experiment. Hybridization was performed as previously described (18). All reactions were performed in duplicate at 70°C. The relative binding ratio (RBR) was calculated by using the method of Brenner et al. (19). The percentage divergence (calculated to the nearest 0.5%) was determined by assuming that each degree of heteroduplex instability, when compared with the melting temperature of the homologous duplex, was caused by 1% unpaired bases (19).
Deep Multilocus Sequencing
Single-end DNA libraries of M. bolletii CIP 108541T, M. chelonae ATCC 35752T, M. immunogenum CIP 106684T, M. massiliense CCUG 48898T, and CV002 were prepared using Illumina DNA Sample Kit (Illumina, Inc., San Diego, CA, USA) according to manufacturer’s recommendations. Sequencing was performed in individual flow cell lanes on the Illumina Genome Analyzer (Illumina, Inc.) at the University of Utah Huntsman Cancer Institute Core Sequencing Facility.
De novo assembly of raw Illumina sequence data was achieved by using Velvet software (20). Velvet was run in 2 parts, velveth and velvetg. For velveth, the hash length was set at 23 (value was selected by calculations in the software manual); default settings were used for all other parameters. In velvetg, the –cov cutoff value was set to auto (setting allows software to automate appropriate coverage cutoff), and –min_contig_lgth was set to 100; all other settings were default.
The genome of M. abscessus CIP 104536T (21) was used as the source reference set of 123 genes that were identified as likely core genome components for the phylum Actinobacter by Ventura et al. (22). The set of reference genes was randomly divided into 5 similarly sized sets to facilitate analysis. SeqMan (DNASTAR Inc., Madison, WI, USA) was used to align the assembled contigs from the sequenced species against each of the sets of reference genes. DNA and inferred amino acid sequences were aligned using MEGA. Only near full-length genes were used in future comparisons. Confirming the translation of the gene was in the correct reading frame relative to the different isolates substantiated quality of each assembled gene. The DNA and amino acid sequences were concatenated for each isolate and sequences alignments and phylogenetic trees were constructed in MEGA using the neighbor-joining method. Kimura 2-parameter distance correction was used for DNA and Poisson correction model was used for amino acid trees; 1,000 bootstrap replications were used for each tree constructed.
Susceptibility Testing
We determined antimicrobial susceptibility by broth microdilution using the recommended Clinical and Laboratory Standards Institute guidelines for rapidly growing Mycobacterium spp (23). Some isolates were not tested for all antimicrobial agents and the concentrations of antimicrobial agents that were tested varied in the panels. MICs of clarithromycin were assessed at 3 days.
Identification of Isolates by Multilocus Sequencing
We obtained 6 type strains representing all members of the M. chelonae-abscessus complex. All type strains and all 127 archived isolates underwent multilocus sequencing. For the 127 archived clinical isolates, we identified 64 M. abscessus, 58 M. chelonae, and 5 M. massiliense isolates. We designated an additional 26 isolates as CV on the basis of our case definition. All 26 CV isolates underwent rpoB gene sequencing, and a subset (n = 11) underwent multilocus sequencing. Unique sequevars are designated among the species M. abscessus, M. massiliense, M. bolletii, M. chelonae, and CV isolates.
16S rRNA Gene
Figure 1

Figure 1. Neighbor-joining tree of a 1,341-bp region of unique 16S rRNA gene sequences of 138 clinical isolates and reference strains of the Mycobacterium chelonae-abscessus complex. Branch support is recorded at nodes as...
Alignments of 1,341 bp of the 16S rRNA gene show 8 unique sequevars from 14 variable bp positions (Figure 1). M. chelonae isolates had the most variability with 4 sequevars and an intraspecies variability of 0.2% (1–3 bp). No M. chelonae clinical isolates shared 100% identity to the type strain ATCC 35758T. M. abscessus isolates had 2 sequevars differing by 1 bp. Two M. massiliense sequevars were observed and differed by 1 bp with 1 sequevar identical to an M. abscessus sequevar. A single sequevar was found for the CV isolates, and was identical to the type strain of M. chelonae.
ITS Region
Figure 2
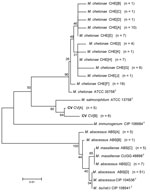
Figure 2. Neighbor-joining tree of 233 bp of unique internal transcribed spacer region sequences of 138 clinical isolates and reference strains of the Mycobacterium chelonae-abscessus complex. Branch support is recorded at nodes as...
Alignments of 233 bp of the ITS region show 18 unique sequevars from 30 variable positions (Figure 2). M. chelonae isolates had the most variability with 12 sequevars and an intraspecies variability of 1.7% (1–4 bp). No M. chelonae clinical isolates shared 100% identity to the type strain ATCC 35758T. M. abscessus isolates showed 4 sequevars with an intraspecies variability of 0.9% (1–2 bp). One M. massiliense sequevar was observed, which was identical to 1 of the M. abscessus sequevars. Two sequevars were found for the CV isolates and differed by 2 bp (0.9%). The CV sequevars were most closely associated with M. salmoniphilum ATCC 13758T at 98.7% (3 bp).
hsp65 Gene
Figure 3
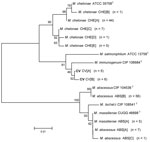
Figure 3. Neighbor-joining tree of a 361-bp region of unique heat-shock protein 65 gene sequences of 138 clinical isolates and reference strains of the Mycobacterium chelonae-abscessus complex. Branch support is recorded at nodes...
Alignments of 361 bp of the hsp65 gene show 11 unique sequevars from 41 variable positions (Figure 3). M. chelonae isolates had the most variability with 5 sequevars and an intraspecies variability of 1.9% (1–7 bp). No M. chelonae clinical isolates shared 100% identity to the type strain ATCC 35758T. M. abscessus isolates had 3 sequevars with an intraspecies variability of 1.7% (1–6 bp). One M. massiliense sequevar was observed and identical to 1 of the M. abscessus sequevars. Two sequevars were found for the CV isolates and differed by 1 bp (0.3%). The CV sequevars were most closely associated with M. immunogenum CIP 106684T at 98.6% (5 bp).
sodA Gene
Figure 4

Figure 4. Neighbor-joining tree of a 426-bp region of unique sodA gene sequences of 138 clinical isolates and reference strains of the Mycobacterium chelonae-abscessus complex. Branch support is recorded at nodes as a...
Alignments of 426 bp of the sodA gene show 15 unique sequevars from 66 variable positions (Figure 4). M. chelonae isolates had the most variability with 9 sequevars and an intraspecies variability of 1.9% (1–8 bp). No M. chelonae clinical isolates shared 100% identity to the type strain ATCC 35758T. M. abscessus isolates had 3 sequevars with an intraspecies variability of 0.7% (1–3 bp). One M. massiliense sequevar was observed, which was identical to 1 of the M. abscessus sequevars. Two sequevars, which differed by 11 bp (2.6%), were found for the CV isolates. The CV sequevars were most closely associated with M. immunogenum CIP 106684T at 95.8% (18 bp).
rpoB Gene
Figure 5

Figure 5. Neighbor-joining tree of a 676-bp region of unique rpoB gene sequences of 153 clinical isolates and reference strains of the Mycobacterium chelonae-abscessus complex. Branch support is recorded at nodes as a...
Alignments of 676 bp of region of the rpoB gene show 19 unique sequevars from 73 variable positions (Figure 5). M. chelonae isolates had the most variability with 9 sequevars and an intraspecies variability of 1.5% (1–10 bp). One M. chelonae clinical isolate shared 100% identity to the type strain ATCC 35758T. M. abscessus isolates had 4 sequevars with an intraspecies variability of 0.7% (1–5 bp). The largest sequevar of M. abscessus clinical isolates had 100% identity to the type strain of M. abscessus. Two M. massiliense sequevars were observed and differed by 2 bp (0.3%). Neither sequevar shared 100% identity with the type strain for M. massiliense. Four sequevars were observed among the CV isolates. They had an interspecies variability of 1.8% (2–11 bp). The CV sequevars were most closely associated with M. chelonae CIP 106684T at 95.1% (33 bp). Sequevar results are summarized in Table 1 in the Technical Appendix .
Deep Multilocus Sequencing
Figure 6
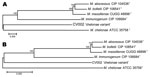
Figure 6. Neighbor-joining tree of DNA (A) and amino acid (B) concatenated gene sequences of Mycobacterium chelonae variant (CV) isolates and reference strains of the M. chelonae-abscessus complex. Branch support is recorded at...
Full-length genes were successfully assembled for 43 genes from 5 type strains and 1 representative clinical CV isolate. Gene names and corresponding GenBank accession numbers are provided in the supplementary tables (Technical Appendix Table 2). Pair-wise alignments of the DNA and amino acid sequences were performed for each isolate (Technical Appendix Tables 3 and 4 ). The 43 concatenated genes ranged from 41,580 to 41,619 bp and created an alignment of 41,792 nt including gaps. Comparisons of DNA percent identity ranged from 89.1% (M. chelonae to M. abscessus) to 98.3% (M. abscessus to M. bolletii). M. massiliense, M. abscessus, and M. bolletii showed a close association with percent identity range of 98.2%–98.3%. The novel CV isolate shared no greater than 90.5% identity with any type strain. Phylogenetic analysis shows closest relationship between M. abscessus, M. massiliense, and M. bolletii (Figure 6, panel A). The CV isolate is on a separate branch with nearly identical distances from M. immunogenum and M. chelonae.
Percent identity of amino acid comparisons ranged from 95.7% (M. chelonae to M. abscessus) to 99.6% (M. abscessus to M. bolletii). M. massiliense, M. abscessus, and M. bolletii showed a close association with percent identity range of 99.5%–99.6%. The novel CV isolate shared no greater than 96.1% identity with any type strain. Phylogenetic analysis shows closest relationship between M. abscessus, M. massiliense, and M. bolletii (Figure 6, panel B).
DNA-DNA Hybridization
DNA-DNA hybridization studies of clinical isolates CV004, CV005, CV006, and CV015 with labeled patient isolate CV002 showed RBRs of 66 to 96% and %D values of 0.0 to 2.0%. The reciprocal DNA-DNA hybridization of patient isolate CV002 with labeled isolate CV005 showed an RBR of 100% and a %D of 0.5%. The DNA-DNA hybridization studies of the phylogenetically (16S rRNA gene) related type strains M. abscessus, M. bolletii, M. chelonae, M. immunogenum, M. massiliense, and M. salmoniphilum performed with the patient isolate CV002 showed RBRs of 15%–69% and %D values of 5.0%–8.0%. DNA-DNA hybridization results are summarized in Technical Appendix Table 5.
Susceptibility Testing
Susceptibilities by broth microdilution were available for most M. chelonae, M. abscessus, M. massiliense, and CV isolates (Table A1). The MICs for the first 3 taxa were comparable those in to previous reports (24). With the exception of 1 isolate, all isolates of M. chelonae were cefoxitin resistant (MIC >128 µg/mL), and M. abscessus and M. massiliense were cefoxitin intermediate (MIC 32–64 µg/mL). All 3 taxa were minocycline resistant. Unlike M. chelonae, CV isolates were intermediate (88%) or susceptible (12%) to cefoxitin.
Clinical Spectrum and Characteristics of CV Isolates
The most common source for the CV isolates was respiratory (n = 20) (Technical Appendix Table 6). The remaining 6 CV isolates had clinical sources that included skin (n = 2), granulomatous liver lesion (n = 1), central line infections (n = 2), and an unspecified body fluid. Most isolates (n = 15) were recovered from patients seen in 4 different hospitals or clinics in Pennsylvania. Eight patients with CV infection were seen at the Hospital of the University of Pennsylvania and 7 of 8 patients were adult females (ages 41–74 years) who acquired the mycobacterial infection as outpatients. Six charts were available for review, and partial information was available for 2 additional patients (patients CV007, CV008, CV010, CV012–CV014, CV034, and CV036).
The medical histories of the 6 patients with complete information fell into 2 groups: those with chronic sinusitis (2 patients) and those with lower respiratory symptoms (4 patients). All patients with lower respiratory symptoms and cultures positive for CV had underlying pulmonary disease (cystic fibrosis, primary ciliary dyskinesia, lung cancer, chronic obstructive pulmonary disease, recurrent pneumonia/bronchiectasis). No patients with lower respiratory symptoms received specific antibiotic therapy aimed at treating rapidly growing mycobacterial infection, although 1 patient received long term antimicrobial drug therapy for concomitant M. avium infection. Two patients with sinusitis were not treated with antimicrobial drugs, but both had sinus surgery with symptomatic improvement. One patient had 3 positive sputum cultures for a rapidly growing Mycobacterium over a 3-year period, 2 of which were shown by genetic sequencing to be CV organisms (CV014 was selected for further study). No other patient had >1 positive culture for CV, if follow-up cultures were performed. Three patients had sequential or concomitant infections with M. abscessus, M. avium-intracellulare, or both bacteria.
The first isolate identified as a CV was discovered in 2005 from a patient in New York. The next isolate was not identified until 2007 from Pennsylvania. Overall, 23 (88%) of 26 were isolated in patients in the northeastern United States, and the remaining 3 isolates were recovered from patients in Minnesota, Oregon, and Colorado.
We describe the discovery of a new human pathogen with clinical features similar to M. abscessus that is implicated as a cause of infection for patients with chronic lung diseases, intravascular catheters, and chronic sinusitis. On the basis of our investigations, we propose CV isolates become a new member of the M. chelonae-abscessus complex and be named Mycobacterium franklinii sp. nov. M. franklinii (frank li′ ni I, N.L. masc. gen. n. franklinii of Franklin, pertaining to Benjamin Franklin, statesman, founder of the University of Pennsylvania, inventor, and scientist who helped create the nation’s first public hospital in Philadelphia, Pennsylvania, USA, the origin of the isolates).
The microorganisms are acid-fast, gram-positive bacilli, and colony morphology alone is not sufficient for differentiation from other rapidly growing Mycobacterium spp. Colonies are nonpigmented appearing on 5% sheep blood agar, Middlebrook 7H10 agar and egg-based Lowenstein-Jensen slants in 2–5 days at temperatures between 24 and 37°C (optimally at 30°C). Even with molecular techniques, underrecognition of this new pathogen is not surprising because it shares 100% full 16S rRNA gene identity with M. chelonae and has sequence variation in the hsp65 gene that results in inconsistent amplification with typical diagnostic primers for sequence or PCR restriction fragment length polymorphism analysis (10,11). However, diagnosis of M. franklinii infection is essential because it is more susceptible to antimicrobial drugs than other members of the M. chelonae-abscessus complex, and its susceptibility pattern with cefoxitin was a distinguishing characteristic leading to its discovery.
Multilocus sequencing on a population of M. chelonae-abscessus complex isolates using 5 DNA regions enabled us to examine a population of closely related isolates to accurately assess species variability. M. franklinii shared complete 16S rRNA gene sequence identity with the type strain of M. chelonae, but was differentiated from M. chelonae and other members of the M. chelonae-abscessus complex by partial sequencing of rpoB, hsp65, sodA, and ITS DNA targets. Concatenated analysis of 43 genes (≈40,000 bp) from deeper sequencing of M. franklinii demonstrated that this novel species shares <90.5% identity with any other M. chelonae-abscessus group member. DNA-DNA hybridization analysis also supports the novel classification with its low relative binding ratios and higher percent divergence from all other M. chelonae-abscessus complex type strains. Cefoxitin susceptibility or intermediate susceptibility is another distinguishing feature. Preliminary testing on 6 M. franklinii isolates revealed inducible resistance (data not shown) in 50% of the isolates upon prolonged clarithromycin incubation (14 days). This finding suggests that similar to M. abscessus, isolates of this species may have an inducible erm gene.
The pathophysiology of diseases associated with M. franklinii is largely unknown. Most M. franklinii isolates were from respiratory sources and from patients with underlying lung conditions. Three of these disorders (cystic fibrosis, primary ciliary dyskinesia, and recurrent pneumonia) had associated bronchiectasis, which is a known risk factor for M. abscessus and M. massiliense nodular lung disease but not for M. chelonae (6,25). It is unclear whether this microorganism causes respiratory tract disease, or simply colonizes damaged airways and sinuses. Similar to patients with M. abscessus and M. chelonae infections, we found 2 cases each of sinusitis and catheter-associated infection from M. franklinii (26). The association with chronic sinusitis presumably relates to sinus washes using tap water rinses in previously diseased sinuses. Although the exact reservoir of M. franklinii is unknown, a recent study in the Netherlands by Van Ingen et al. reported 2 isolates from tap and shower water based on rpoB gene sequence (27), and these 2 isolates shared 99.6% identity to our M. franklinii sequevar A rpoB gene sequence. The observation in the Netherlands suggests an environmental source for this organism, and it is likely that the novel species is regionally specific and can survive in municipal water sources. This hypothesis would be supported by the large number of cases in a focused region in Pennsylvania.
Our population analyses of clinical isolates of M. chelonae demonstrate a lack of taxonomic clarity. Additionally, our investigations lend further evidence that species distinctions for M. bolletii and M. massiliense may be inappropriate and support the recent proposal to modify their classifications (5,28). Taxonomic uncertainty likely arises as our understanding of microbial phylogeny expands with rapid advances in technologies and often results in inconsistent standards being applied for species designations. For example, DNA-DNA hybridization is a relatively standard technique, but upon review of 14 species descriptions in 2009 only 5 were supported by using DNA-DNA hybridization (29–36).
The discovery of an emerging pathogen should be taken in the context of microbial ecology and evolution, the interaction between host and microbe, and factors of virulence. This investigation underscores the need for accurate identification of Mycobacterium spp. for detection of a new pathogen. The interplay between colonization and disease is not clearly defined, but we demonstrate its role in central line infections and for patients with sinopulmonary disease. M. franklinii may have newly emerged as a human pathogen over the past 5 years, or it has been involved in human disease previously and was unrecognized. In order to further our understanding of this pathogen and its role in disease, greater surveillance and awareness is necessary. At this time, clinical laboratories can identify M. franklinii by sequencing based assays that target either the ITS region (between 16S and 23S rRNA genes), hsp65, rpoB, and sodA genes, or by complete 16S rRNA gene sequence analysis in conjunction with cefoxitin and minocycline susceptibility patterns. The type strain, CV002 (ATCC [pending] and DSMZ 45524), was isolated from a skin lesion.
Mr Simmon is a research scientist and bioinformaticist at Associated Regional and University Pathologists Laboratories. His research focuses on diagnostic testing for infectious diseases and disease surveillance.
Acknowledgment
We dedicate this manuscript to medical technologists whose astute observations often serve as catalysts for our work. In particular, we thank Linda Bridge Mann, Rebecca Wilson, Ravikan Vasireddy, Steven McNulty, and Maria McGlasson and members of the Molecular Pathology and Clinical Microbiology Laboratories at the Hospital of the University of Pennsylvania and Associated Regional and University Pathologists Laboratories. Additionally, we thank Paul Bourbeau, Karen Brady, Christine Turenne, and Hans Truper for their assistance and helpful discussions.
References
- Wallace RJ Jr, Zhang Y, Wilson RW, Mann L, Rossmoore H. Presence of a single genotype of the newly described species Mycobacterium immunogenum in industrial metalworking fluids associated with hypersensitivity pneumonitis. Appl Environ Microbiol. 2002;68:5580–4. DOIPubMedGoogle Scholar
- Adékambi T, Reynaud-Gaubert M, Greub G, Gevaudan MJ, La Scola B, Raoult D, Amoebal coculture of “Mycobacterium massiliense” sp. nov. from the sputum of a patient with hemoptoic pneumonia. J Clin Microbiol. 2004;42:5493–501. DOIPubMedGoogle Scholar
- Adékambi T, Berger P, Raoult D, Drancourt M. rpoB gene sequence-based characterization of emerging non-tuberculous mycobacteria with descriptions of Mycobacterium bolletii sp. nov., Mycobacterium phocaicum sp. nov. and Mycobacterium aubagnense sp. nov. Int J Syst Evol Microbiol. 2006;56:133–43. DOIPubMedGoogle Scholar
- Whipps CM, Butler WR, Pourahmad F, Watral VG, Kent ML. Molecular systematics support the revival of Mycobacterium salmoniphilum (ex Ross 1960) sp. nov., nom. rev., a species closely related to Mycobacterium chelonae. Int J Syst Evol Microbiol. 2007;57:2525–31. DOIPubMedGoogle Scholar
- Leao SC, Tortoli E, Euzéby JP, Garcia MJ. Proposal that the two species Mycobacterium massiliense and Mycobacterium bolletii be reclassified as Mycobacterium abscessus subsp. bolletii comb. nov., designation of Mycobacterium abscessus subsp. abscessus subsp. nov., and emendation of Mycobacterium abscessus. Int J Syst Evol Microbiol. 2010; [Epub ahead of print].
- Griffith DE, Girard WM, Wallace RJ Jr. Clinical features of pulmonary disease caused by rapidly growing mycobacteria. An analysis of 154 patients. Am Rev Respir Dis. 1993;147:1271–8.PubMedGoogle Scholar
- Wallace RJ Jr, Swenson JM, Silcox VA, Good RC, Tschen JA, Stone MS. Spectrum of disease due to rapidly growing mycobacteria. Rev Infect Dis. 1983;5:657–79. DOIPubMedGoogle Scholar
- Simmon KE, Pounder JI, Greene JN, Walsh F, Anderson CM, Cohen S, Identification of an emerging pathogen, Mycobacterium massiliense, by rpoB sequencing of clinical isolates collected in the United States. J Clin Microbiol. 2007;45:1978–80. DOIPubMedGoogle Scholar
- Cloud JL, Hoggan K, Belousov E, Cohen S, Brown-Elliott BA, Mann L, Use of the MGB Eclipse system and SmartCycler PCR for differentiation of Mycobacterium chelonae and M. abscessus. J Clin Microbiol. 2005;43:4205–7. DOIPubMedGoogle Scholar
- Steingrube VA, Gibson JL, Brown BA, Zhang Y, Wilson RW, Rajagopalan M, PCR amplification and restriction endonuclease analysis of a 65-kilodalton heat shock protein gene sequence for taxonomic separation of rapidly growing mycobacteria. J Clin Microbiol. 1995;33:149–53.PubMedGoogle Scholar
- Telenti A, Marchesi F, Balz M, Bally F, Bottger EC, Bodmer T. Rapid identification of mycobacteria to the species level by polymerase chain reaction and restriction enzyme analysis. J Clin Microbiol. 1993;31:175–8.PubMedGoogle Scholar
- Adékambi T, Colson P, Drancourt M. rpoB-based identification of nonpigmented and late-pigmenting rapidly growing mycobacteria. J Clin Microbiol. 2003;41:5699–708. DOIPubMedGoogle Scholar
- Roth A, Reischl U, Streubel A, Naumann L, Kroppenstedt RM, Habicht M, Novel diagnostic algorithm for identification of mycobacteria using genus-specific amplification of the 16S-23S rRNA gene spacer and restriction endonucleases. J Clin Microbiol. 2000;38:1094–104.PubMedGoogle Scholar
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–9. DOIPubMedGoogle Scholar
- Adékambi T, Drancourt M. Dissection of phylogenetic relationships among 19 rapidly growing Mycobacterium species by 16S rRNA, hsp65, sodA, recA and rpoB gene sequencing. Int J Syst Evol Microbiol. 2004;54:2095–105. DOIPubMedGoogle Scholar
- Loeffelholz MJ, Scholl DR. Method for improved extraction of DNA from Nocardia asteroides. J Clin Microbiol. 1989;27:1880–1.PubMedGoogle Scholar
- Wayne LG. International Committee on Systematic Bacteriology: announcement of the report of the ad hoc Committee on Reconciliation of Approaches to Bacterial Systematics. Zentralbl Bakteriol Mikrobiol Hyg [A]. 1988;268:433–4.PubMedGoogle Scholar
- Brenner DJ, McWhorter AC, Knutson JK, Steigerwalt AG. Escherichia vulneris: a new species of Enterobacteriaceae associated with human wounds. J Clin Microbiol. 1982;15:1133–40.PubMedGoogle Scholar
- Brenner DJ, Hickman-Brenner FW, Lee JV, Steigerwalt AG, Fanning GR, Hollis DG, Vibrio furnissii (formerly aerogenic biogroup of Vibrio fluvialis), a new species isolated from human feces and the environment. J Clin Microbiol. 1983;18:816–24.PubMedGoogle Scholar
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9. DOIPubMedGoogle Scholar
- Ripoll F, Pasek S, Schenowitz C, Dossat C, Barbe V, Rottman M, Non mycobacterial virulence genes in the genome of the emerging pathogen Mycobacterium abscessus. PLoS ONE. 2009;4:e5660. DOIPubMedGoogle Scholar
- Ventura M, Canchaya C, Tauch A, Chandra G, Fitzgerald GF, Chater KF, Genomics of Actinobacteria: tracing the evolutionary history of an ancient phylum. Microbiol Mol Biol Rev. 2007;71:495–548. DOIPubMedGoogle Scholar
- Woods GL, Brown-Elliott BA, Desmond EP, Hall GS, Heifets L, Pfyffer GE, Susceptibility testing of mycobacteria, nocardiae, and other aerobic actinomycetes; approved standard. National Committee on Clinical and Laboratory Standards;23:M24-A. Wayne (PA): The Committee; 2003.
- Swenson JM, Wallace RJ Jr, Silcox VA, Thornsberry C. Antimicrobial susceptibility of five subgroups of Mycobacterium fortuitum and Mycobacterium chelonae. Antimicrob Agents Chemother. 1985;28:807–11.PubMedGoogle Scholar
- Roux AL, Catherinot E, Ripoll F, Soismier N, Macheras E, Ravilly S, Multicenter study of prevalence of nontuberculous mycobacteria in patients with cystic fibrosis in France. J Clin Microbiol. 2009;47:4124–8. DOIPubMedGoogle Scholar
- Redelman-Sidi G, Sepkowitz KA. Rapidly growing mycobacteria infection in patients with cancer. Clin Infect Dis. 2010;51:422–34. DOIPubMedGoogle Scholar
- van Ingen J, Blaak H, de Beer J, de Roda Husman AM, van Soolingen D. Rapidly growing nontuberculous mycobacteria cultured from home tap and shower water. Appl Environ Microbiol. 2010;76:6017–9. DOIPubMedGoogle Scholar
- Leao SC, Tortoli E, Viana-Niero C, Ueki SY, Lima KV, Lopes ML, Characterization of mycobacteria from a major Brazilian outbreak suggests that revision of the taxonomic status of members of the Mycobacterium chelonae-M. abscessus group is needed. J Clin Microbiol. 2009;47:2691–8. DOIPubMedGoogle Scholar
- Ben Salah I, Cayrou C, Raoult D, Drancourt M. Mycobacterium marseillense sp. nov., Mycobacterium timonense sp. nov. and Mycobacterium bouchedurhonense sp. nov., members of the Mycobacterium avium complex. Int J Syst Evol Microbiol. 2009;59:2803–8. DOIPubMedGoogle Scholar
- Hennessee CT, Seo JS, Alvarez AM, Li QX. Polycyclic aromatic hydrocarbon-degrading species isolated from Hawaiian soils: Mycobacterium crocinum sp. nov., Mycobacterium pallens sp. nov., Mycobacterium rutilum sp. nov., Mycobacterium rufum sp. nov. and Mycobacterium aromaticivorans sp. nov. Int J Syst Evol Microbiol. 2009;59:378–87. DOIPubMedGoogle Scholar
- Okazaki M, Ohkusu K, Hata H, Ohnishi H, Sugahara K, Kawamura C, Mycobacterium kyorinense sp. nov., a novel, slow-growing species, related to Mycobacterium celatum, isolated from human clinical specimens. Int J Syst Evol Microbiol. 2009;59:1336–41. DOIPubMedGoogle Scholar
- Tortoli E, Baruzzo S, Heijdra Y, Klenk HP, Lauria S, Mariottini A, Mycobacterium insubricum sp. nov. Int J Syst Evol Microbiol. 2009;59:1518–23. DOIPubMedGoogle Scholar
- van Ingen J, Al-Hajoj SA, Boeree M, Al-Rabiah F, Enaimi M, de Zwaan R, Mycobacterium riyadhense sp. nov., a non-tuberculous species identified as Mycobacterium tuberculosis complex by a commercial line-probe assay. Int J Syst Evol Microbiol. 2009;59:1049–53. DOIPubMedGoogle Scholar
- van Ingen J, Boeree MJ, de Lange WC, de Haas PE, van der Zanden AG, Mijs W, Mycobacterium noviomagense sp. nov.; clinical relevance evaluated in 17 patients. Int J Syst Evol Microbiol. 2009;59:845–9. DOIPubMedGoogle Scholar
- van Ingen J, Boeree MJ, Kosters K, Wieland A, Tortoli E, Dekhuijzen PN, Proposal to elevate Mycobacterium avium complex ITS sequevar MAC-Q to Mycobacterium vulneris sp. nov. Int J Syst Evol Microbiol. 2009;59:2277–82. DOIPubMedGoogle Scholar
- van Ingen J, Lindeboom JA, Hartwig NG, de Zwaan R, Tortoli E, Dekhuijzen PN, Mycobacterium mantenii sp. nov., a pathogenic, slowly growing, scotochromogenic species. Int J Syst Evol Microbiol. 2009;59:2782–7. DOIPubMedGoogle Scholar
Figures
Table
Follow Up
Earning MEDSCAPE CME Credit
To obtain credit, you should first read the journal article. After reading the article, you should be able to answer the following, related, multiple-choice questions. To complete the questions (with a minimum 70% passing score) and earn continuing medical education (CME) credit, please go to www.medscape.org/journal/eid. Credit cannot be obtained for tests completed on paper, although you may use the worksheet below to keep a record of your answers. You must be a registered user on Medscape.org. If you are not registered on Medscape.org, please click on the New Users: Free Registration link on the left hand side of the website to register. Only one answer is correct for each question. Once you successfully answer all post-test questions you will be able to view and/or print your certificate. For questions regarding the content of this activity, contact the accredited provider, CME@medscape.net. For technical assistance, contact CME@webmd.net. American Medical Association’s Physician’s Recognition Award (AMA PRA) credits are accepted in the US as evidence of participation in CME activities. For further information on this award, please refer to http://www.ama-assn.org/ama/pub/category/2922.html. The AMA has determined that physicians not licensed in the US who participate in this CME activity are eligible for AMA PRA Category 1 Credits™. Through agreements that the AMA has made with agencies in some countries, AMA PRA credit may be acceptable as evidence of participation in CME activities. If you are not licensed in the US, please complete the questions online, print the certificate and present it to your national medical association for review.
Article Title: Mycobacterium chelonae-abscessus Complex Associated with Sinopulmonary Disease, Northeastern USA
CME Questions
1. Which of the following statements regarding the members of the Mycobacterium chelonae-abscessus complex is most accurate?
A. There are 22 known members of the M. chelonae-abscessus complex
B. These mycobacteria can promote skin infections, pneumonia, and abscesses
C. Infections occur only among immunocompromised hosts
D. There are clear taxonomical relationships between members of the M. chelonae-abscessus complex
2. "Mycobacterium franklinii" shared which of the following gene sequences with M. chelonae in the current study?
A. rpoB
B. sodA
C. hsp65
D. 16S rRNA
3. What was the most common source of "M. franklinii" in the current study?
A. Skin
B. Liver lesion
C. Central line
D. Respiratory
4. Which of the following statements regarding the antimicrobial susceptibility of "M. franklinii" is most accurate?
A. "M. franklinii" has the same susceptibility profile as other members of the M. chelonae-abscessus complex
B. "M. franklinii " is more likely to be susceptible to minocycline compared with other members of the M. chelonae-abscessus complex
C. "M. franklinii " is more likely to be susceptible to cefoxitin compared with other members of the M. chelonae-abscessus complex
D. "M. franklinii " is more likely to be resistant to cefoxitin compared with other members of the M. chelonae-abscessus complex
Activity Evaluation
|
1. The activity supported the learning objectives. |
||||
|
Strongly Disagree |
|
|
|
Strongly Agree |
|
1 |
2 |
3 |
4 |
5 |
|
2. The material was organized clearly for learning to occur. |
||||
|
Strongly Disagree |
|
|
|
Strongly Agree |
|
1 |
2 |
3 |
4 |
5 |
|
3. The content learned from this activity will impact my practice. |
||||
|
Strongly Disagree |
|
|
|
Strongly Agree |
|
1 |
2 |
3 |
4 |
5 |
|
4. The activity was presented objectively and free of commercial bias. |
||||
|
Strongly Disagree |
|
|
|
Strongly Agree |
|
1 |
2 |
3 |
4 |
5 |
1These authors contributed equally to this article.
Related Links
Table of Contents – Volume 17, Number 9—September 2011
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Keith E. Simmon, Associated Regional and University Pathologists Laboratories, 500 Chipeta Way, Salt Lake City, UT 84108, USA
Top