Volume 19, Number 4—April 2013
Dispatch
Hepatitis Virus in Long-Fingered Bats, Myanmar
Figure 1
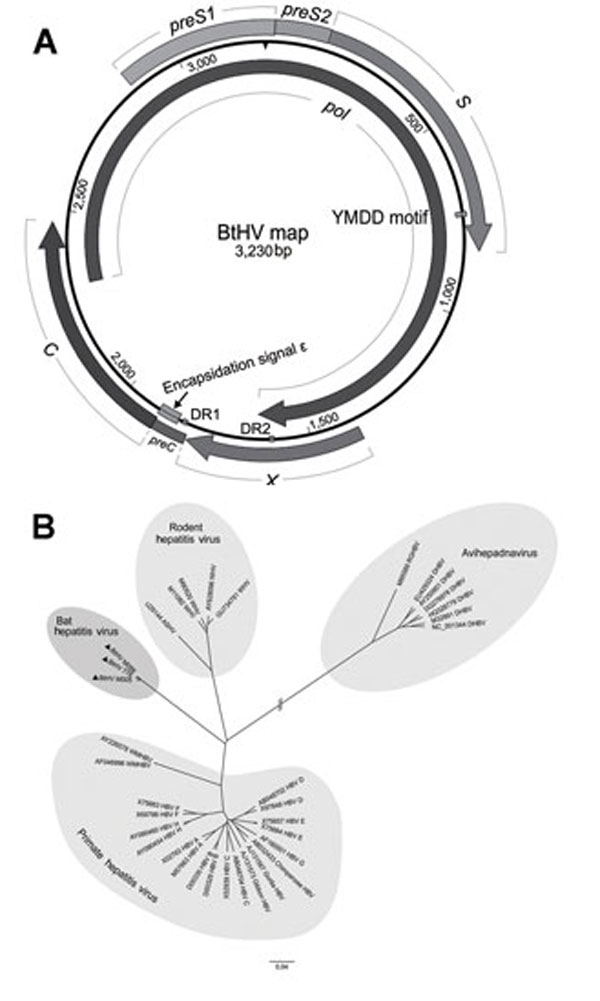
Figure 1. . Predicted schematic representation of the bat hepatitis virus (BtHV) genome and its phylogenetic relationship with other hepadnaviruses. A) Genomic structural map of BtHV. Boxes and arrows represent the open reading frames encoding the main proteins: pol gene (2,305–1,636), preS1/S2 and S gene (2,864–833), preC/C gene (1,815–2,468) and X gene (1,378–1,812). Two 12-nt direct repeat sequences (DR1 from 1,825 to 1,836 and DR2 from 1,594 to 1,605), the encapsidation signal ε (1,848–1,903), and YMDD domain (734–745) are also depicted in the map. B) Phylogenetic analysis of BtHVs and other hepadnaviruses based on amino acid sequences of pol genes. Representatives of hepadnavirus species belonging to Orthohepadnavirus and Avihepadnavirus genera were used; their GenBank accession nos. are shown in the trees. The different genotypes of human hepatitis B virus are also included. The 3 BtHV isolates are identified by black triangles. Scale bar indicates nucleotide substitutions per site.
1These authors contributed equally to this article.