Volume 20, Number 1—January 2014
Dispatch
Rapidly Fatal Hemorrhagic Pneumonia and Group A Streptococcus Serotype M1
Cite This Article
Citation for Media
Abstract
We report 3 cases of fulminant hemorrhagic pneumonia in previously health patients. Sudden-onset hemoptysis and dyspnea developed; all 3 patients and died <12 h later of massive pulmonary bleeding, despite aggressive supportive care. Postmortem analysis showed that the illnesses were caused by group A Streptococcus emm1/sequence type 28 strains.
Streptococcus pyogenes or group A Streptococcus (GAS) is a versatile pathogen that can cause serious diseases, including bacteremia, cellulitis, puerperal sepsis, meningitis, pneumonia, and necrotizing fasciitis (1,2). This pathogen also causes streptococcal toxic shock syndrome, a severe response to streptococcal pyrogenic exotoxins (Spe proteins), which trigger overproduction of inflammatory cytokines, leading to tissue damage, organ failure, and shock (3,4). Despite improved awareness and treatment, GAS infections remain among the top 10 infectious causes of fatal disease in humans (1).
Periodic resurgences of invasive GAS infections in industrialized countries have been linked to emergence of dominant GAS clones resulting from horizontal gene transfer (5–8). The ability of these clones to cause life-threatening disease in healthy persons probably depends on expression of virulence factors that facilitate penetration of host cell barriers and evasion of immune defenses (3,4,8).
More than 250 GAS types have been identified in sequencing studies of the hypervariable region of the emm gene, which encodes the M surface protein (1,2). The emm types seem to correlate with tissue tropism of the organism, and M protein augments GAS virulence by interfering with antibody and complement deposition, facilitating formation of microcolonies and neutralizing antimicrobial peptides, and stimulating proinflammatory and procoagulatory activities (3,4). M1 protein and M1 fragments released by neutrophil proteases can also provoke pulmonary hemorrhage, inflammation, and tissue destruction by their interaction with fibrinogen, which causes neutrophils to release heparin-binding protein, a mediator of vascular leakage (9). The hypervirulent emm1 GAS clone also harbors prophages encoding SpeA proteins and extracellular streptodornase D (Sda1), which can also enhance virulence and dissemination (8,10). We report 3 cases of rapidly fatal, hemorrhagic pneumonia in previously healthy patients that were determined to be caused by infection with hypervirulent GAS.
Patient 1 was a 74-year-old Italian man who came to the emergency department (ED) of Vittorio Emanuele Hospital in Catania, Italy, in July 2012. Patient 2 was 65-year-old German woman who arrived in Rome from Frankfurt in March 2011 and came directly from the airport to the ED of the University of Rome Medical Center. Patient 3 was a 40-year-old Japanese man who came to the ED of the Catholic University Medical Center in Rome in February 2012, 3 days after arriving in the city. All 3 patients had sudden-onset hemoptysis, dyspnea, and fever (temperature 37.5–38.5°C), which had rapidly worsened over 2–3 h. Their medical histories were unremarkable.
Figure
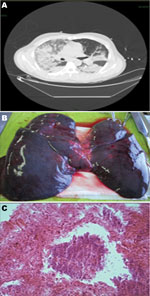
Figure. . A) Computed tomographic image of the chest of a 74-year-old patient (patient 1) with fatal hemorrhagic pneumonia, Catania, Italy, showing multifocal confluent parenchymal opacities. B) Postmortem view of the lungs...
All 3 patients had acute respiratory failure requiring ventilator support and hemoconcentration; hyponatremia; increased levels of serum creatinine, lactate dehydrogenase, lactic acid, and brain natriuretic peptides; leukocytosis (7,000–8,000 cells/mm3, 3,200–4,000 neutrophils); and increased levels of C-reactive protein (>400 mg/mL) and
Autopsy specimens showed bilateral hemorrhagic pleural effusions (1,500–2,000 cells/mL), heavy, blood-engorged lungs (1,600–1,700 g), and patent hilar structures, but no thoracic or abdominal lymphadenopathy. Lungs showed hemorrhage and edema (Figure, panel B). Microscopic analysis of the lungs showed necrosis and bacteria (Figure, panel C). Admission blood cultures (3 sets/patient) were positive at the 12-h reading. S. pyogenes was identified by using matrix-assisted laser desorption ionization–time-of-flight mass spectrometry (Bruker Daltonik, Breman, Germany) and isolated in all culture bottles, bronchoalveolar lavage cultures (pure colonies, 106 CFU/mL), and postmortem lung tissue cultures.
All isolates were susceptible to erythromycin, tetracycline, amoxicillin, penicillin, and clindamycin by Etest (bioMérieux, Marcy l’Etoile, France). Results were interpreted according to European Committee on Antimicrobial Susceptibility Testing breakpoints (www.eucast.org/clinical_breakpoints). Results of testing for urinary Legionella pneumophila and S. pneumoniae antigens, β-glucan, galactomannan, and HIV and toxicology panels were negative for all 3 patients. Commercially available PCRs for respiratory tract samples showed negative results for major respiratory viruses and bacterial pathogens. Antibody titers against streptolysin O were <200 IU.
The emm typing, which was performed by PCR using protocols and the database of the Centers for Disease Control and Prevention (Atlanta, GA, USA; www.cdc.gov/ncidod/biotech/strep/M-ProteinGene_typing.htm), showed that all 3 GAS strains were emm-type 1 and had identical sequences at the 5ʹ end of the emm gene, indicative of the emm 1.0 allele. Strains were tested for multiple virulence genes (Table) by using PCR and primers described elsewhere (11) or designed with the VectorNTI program (Invitrogen, Carlsbad, CA, USA). They were identical in terms of the spe genotype (speA+, speB+, speC–, speG+, speI–, speJ+, smeZ+, ssa–); the presence of SLO, sagA, sagBC, and sda1 genes; and the absence of PAM, prtF, and sof genes. Multilocus sequence typing was performed as described (http://spyogenes.mlst.net/) and showed an identical sequence type (ST) (ST28/CC28) for the isolates from patients 1 and 3 and a new single-locus variant of ST28 (designated ST648) for the isolate from patient 2. All cases were caused by GAS strains harboring the emm1.0 allele and Sda1, smeZ, speA, speB, speG, and speJ genes.
A total of 1.3%–23.8% invasive GAS infections involve pneumonia, but these infections are more common in developing countries (1,6,7,12). Successful management depends on early diagnosis and initiation of effective treatment (2,3). However, despite early aggressive supportive care and empirical antimicrobial drug therapy, which was later confirmed to be appropriate by antibiogram results, all 3 patients we describe died within 8–12 h of symptom onset from massive pulmonary bleeding and acute respiratory failure. Initial presentations were fairly nonspecific, and diagnoses were made postmortem. The roles of GAS in the rapid progression of disease were highlighted by the abundance of GAS found the lungs, extensive and severe pulmonary damage, and virulence factor profiles of the isolates, all of which included sda1, smeZ, speA, speB, speG, speJ, and emm-1 genes (4,5).
Invasive bacterial disease requires virulence factors that facilitate interactions of the microbe with host tissues and subvert defenses of the immune system. In the emm1 GAS clone, progression to systemic infection is also favored by mutations in the 2-component control of virulence regulatory system, which enhances resistance to subepithelial immune defenses and facilitates deep-tissue penetration. These mutations markedly alter transcription profiles of invasive GAS isolates than those of pharyngeal mucosal isolates (3–5,8), strongly upregulating sda1 transcription and markedly downregulating expression of the gene encoding cysteine protease SpeB (6–12). The sda1 gene facilitates avoidance of neutrophil extracellular traps by the pathogen and serves as a selective force for a control of virulence regulatory system mutation (7,13). SpeB protease enables accumulation and activation of broad-spectrum host protease plasmin on the microbial cell surface, thereby promoting infection spread to normally sterile sites (2,3,14). Invasive GAS strains also produce increased levels of toxins, including some that destroy immune cells, and superantigens (e.g., SpeA, SpeJ) that dysregulate the immune response of the host (3).
Host factors also affect clinical presentation and disease progression, which explains why diseases of different severity can be caused by genetically indistinguishable emm1 strains with no evidence of regulatory gene mutation (2,4,7). Elucidation of these host and bacterial factors involved in the pathogenesis of these rare but life-threatening infections may be useful for improving disease prognosis (6).
Dr Santagati is a research scientist in microbiology at the University of Catania, Catania, Italy. Her research interests include virulence mechanisms of gram-positive bacteria, characterization of genetic elements associated with antibiotic resistance genes, and gram-positive bacteriocin producers and their clinical application.
Acknowledgments
We thank Marian Everett Kent for assistance in editing the manuscript.
This study was supported by the Ministero dell’Istruzione, dell’Università e della Ricerca (grant 2012 UNICT to S.S).
References
- Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005;5:685–94. DOIPubMedGoogle Scholar
- Steer AC, Lamagni T, Curtis N, Carapetis JR. Invasive group a streptococcal disease: epidemiology, pathogenesis and management. Drugs. 2012;72:1213–27. DOIPubMedGoogle Scholar
- Cole JN, Barnett TC, Nizet V, Walker MJ. Molecular insight into invasive group A streptococcal disease. Nat Rev Microbiol. 2011;9:724–36. DOIPubMedGoogle Scholar
- Henningham A, Barnett TC, Maamary PG, Walker MJ. Pathogenesis of group A streptococcal infections. Discov Med. 2012;13:329–42 .PubMedGoogle Scholar
- Friães A, Pinto FR, Silva-Costa C, Ramirez M, Melo-Cristino J. Group A streptococci clones associated with invasive infections and pharyngitis in Portugal present differences in emm types, superantigen gene content and antimicrobial resistance. BMC Microbiol. 2012;12:280. DOIPubMedGoogle Scholar
- Lamagni TL, Efstratiou A, Vuopio-Varkila J, Jasir A, Schalén C. The epidemiology of severe Streptococcus pyogenes associated disease in Europe. Euro Surveill. 2005;10:179–84 .PubMedGoogle Scholar
- Turner CE, Dryden M, Holden MT, Davies FJ, Lawrenson RA, Farzaneh L, Molecular analysis of an outbreak of lethal postpartum sepsis caused by Streptococcus pyogenes. J Clin Microbiol. 2013;51:2089–95. DOIPubMedGoogle Scholar
- Sumby P, Porcella SF, Madrigal AG, Barbian KD, Virtaneva K, Ricklefs SM, Evolutionary origin and emergence of a highly successful clone of serotype M1 group A Streptococcus involved multiple horizontal gene transfer events. J Infect Dis. 2005;192:771–82. DOIPubMedGoogle Scholar
- Herwald H, Cramer H, Mörgelin M, Russell W, Sollenberg U, Norrby-Teglund A, M protein, a classical bacterial virulence determinant, forms complexes with fibrinogen that induce vascular leakage. Cell. 2004;116:367–79. DOIPubMedGoogle Scholar
- Uchiyama S, Andreoni F, Schuepbach RA, Nizet V, Zinkernagel AS. DNase Sda1 allows invasive M1T1 group A Streptococcus to prevent TLR9-dependent recognition. PLoS Pathog. 2012;8:e1002736. DOIPubMedGoogle Scholar
- Santagati M, Scillato M, Patane F, Aiello C, Stefani S. Bacteriocin-producing oral streptococci and inhibition of respiratory pathogens. FEMS Immunol Med Microbiol. 2012;65:23–31. DOIPubMedGoogle Scholar
- Lassalle S, Hofman V, Butori C, Sicard D, Hofman P. Histological diagnosis of lung bacterial necrotizing infections: a study of four fulminant cases. Ann Pathol. 2005;25:349–56. DOIPubMedGoogle Scholar
- Walker MJ, Hollands A, Sanderson-Smith ML, Cole JN, Kirk JK, Henningham A, DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat Med. 2007;13:981–5. DOIPubMedGoogle Scholar
- Kansal RG, McGeer A, Low DE, Norrby-Teglund A, Kotb M. Inverse relation between disease severity and expression of the streptococcal cysteine protease, SpeB, among clonal M1T1 isolates recovered from invasive group A streptococcal infection cases. Infect Immun. 2000;68:6362–9 . DOIPubMedGoogle Scholar
Figure
Table
Cite This Article1These authors contributed equally to this article.
Table of Contents – Volume 20, Number 1—January 2014
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Stefania Stefani, Department of Bio-medical Science, Section of Microbiology, University of Catania, Via Androne 81, 95124 Catania, ItalyStefania Stefani, Department of Bio-medical Science, Section of Microbiology, University of Catania, Via Androne 81, 95124 Catania, Italy
Top