Volume 20, Number 2—February 2014
Dispatch
Co-circulation of West Nile Virus Variants, Arizona, USA, 2010
Cite This Article
Citation for Media
Abstract
Molecular analysis of West Nile virus (WNV) isolates obtained during a 2010 outbreak in Maricopa County, Arizona, USA, demonstrated co-circulation of 3 distinct genetic variants, including strains with novel envelope protein mutations. These results highlight the continuing evolution of WNV in North America and the current complexity of WNV dispersal and transmission.
West Nile virus (WNV) emerged in the Americas in 1999 after an outbreak of neuroinvasive disease in humans, birds, and horses in New York, New York. The virus spread rapidly across North America and was detected in Arizona in 2003. In 2004, Arizona experienced a large outbreak (214 neuroinvasive cases and 16 deaths, second only to California in that year), followed by ≈50–60 neuroinvasive cases per year during 2005–2008.
An outbreak in 2010 resulted in 107 neuroinvasive cases and 15 deaths, the largest number of cases for a state that year. WNV activity in Maricopa County, which includes the city of Phoenix and surrounding municipalities, where numerous human cases were reported in the town of Gilbert, was investigated by a team from the Centers for Disease Control and Prevention (Fort Collins, CO, USA) working with local and state public health officials. Epidemiologic and entomologic findings from those investigations have been reported (1,2) We describe the molecular and phenotypic characterization of WNV isolates obtained from that outbreak.
Figure 1
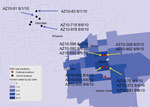
Figure 1. . Distribution of mosquito sampling sites in Maricopa County, Arizona, USA, during the 2010 West Nile virus (WNV) outbreak investigation and collection dates/locations of pools yielding indicated WNV isolates used for...
Figure 2
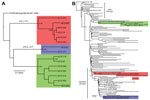
Figure 2. . A) Bayesian phylogenetic tree of envelopes genes of all described Arizona, USA, 2010 isolates of West Nile virus (WNV) (n = 15). Isolates grouped in 3 distinct monophyletic clusters designated...
As part of the Centers for Disease Control and Prevention investigation, Vero cell culture isolates of WNV were obtained from pools of Culex quinquefasciatus (Say) mosquitoes collected during August 1–9, 2010, at multiple sites in the study areas (Figure 1). Nucleotide sequences (GenBank accession nos. KF704145–KF704159) for the envelope (E) protein–coding regions were determined for 15 strains and subjected to phylogenetic analysis. The AZ10 E gene sequences were distributed in 3 robust monophyletic clusters (designated A, B, C; posterior probabilities 0.99) as determined by using applied relaxed clock Bayesian coalescent analysis (Figure 2, panel A).
Isolates from all 3 clusters were detected from the Gilbert study sites. All AZ10 isolates encoded the E-159 Val→Ala mutation that is characteristic of genotypes described since 2002 (4). The 7 strains in cluster A, which were only obtained from pools collected in Gilbert, also encoded a conservative Leu→Ile mutation at E-312, a surface exposed residue in the putative receptor binding domain III (EIII) known to be a variable site in multiple WNV genetic lineages, including strains of lineage 2 currently circulating in Europe (5–9). The 2 strains in cluster B, collected in Glendale and Gilbert, Arizona, encoded a conservative Ser→Thr mutation at E-275.
To further characterize the phylogenetic relationships of these AZ10 isolates, 1 strain from each cluster was selected for full-length genomic sequencing and comparison of the encoded open reading frames to 486 additional genomic sequences from North America available in GenBank. This analysis also supported the concurrent circulation of 3 distinct variants in Gilbert and the surrounding areas of Maricopa County during the 2010 outbreak (Figure 2, panel B). Strain AZ10–581 (cluster A) grouped with the recently described SW/WN03 genotype (10), and was most closely related to a South Dakota 2005 strain and 2 other strains that each encoded the E-L312I mutation. Strain AZ10–91 (cluster B) grouped with 2004–2005 Arizona and New Mexico isolates also belonging to a clade of the SW/WN03 genotype. Other SW/WN03 genotype viruses did not encode the E-275 mutation in AZ10–91 and AZ10–372. Strain AZ10–892 grouped with other recently described Arizona 2010/2011 isolates (4) and a New York 2004 strain belonging to the dominant NA/WN02 genotype, confirming persistence or reintroduction of that genotype in the southwestern United States (4). Nucleotide divergence from NY99 ranged from 0.58% to 0.66% for the AZ10 strains and divergence between the 3 clusters was up to ≈1.2% (Table 1).
The presence of the E-312 coding mutation was of particular interest. Most sequences for lineage 1 WNV strains encode Leu at E-312, whereas lineage 2 strains encode Val or Ala. E-312 lies in an exposed loop of EIII, where it may contribute to the antigenic and/or putative receptor binding activities of the domain (6). To assess the effects of the Leu→Ile mutation and tolerance for alternative amino acid substitutions at this site, we engineered 5 E-312 mutants by using an NY99 infectious clone (NY99ic) encoding alternative amino acids that are each only a single nucleotide substitution away from the wild-type Leu codon (CUU) (Table 2). (Although Phe also requires only a single nucleotide change from the Leu codon, it occurs naturally in some lineage 1 and 2 WNV strains and was not included in this analysis.) Mutagenesis, in vitro ligation, and transcription of genome equivalent RNA and virus recovery were performed as described (11). All mutant viruses were readily recovered from transfected Vero cells and grew to peak titers comparable to the parental NY99ic virus, and the introduced mutations were stable through 3 additional Vero cell passages.
Virulence of AZ10-75 and AZ10−581 (cluster A), AZ10-91 (cluster B), AZ10-892 (cluster C), and the recovered NY99ic E-312 mutants was compared with wild-type NY99ic after intraperitoneal inoculation of 3- to 4-week-old female Swiss Webster mice (Table 2) as described (11). The AZ10 strains and all E-312 mutants had 50% lethal doses (LD50s) and average survival times comparable with that of NY99ic, with the exception of the L312P mutant, which was markedly attenuated (630 PFU/LD50 vs. 0.3 PFU/LD50, and prolonged survival time). Antigenic characteristics of viruses encoding L312I mutations were also compared by assessing their neutralization by monoclonal antibodies 7H2 and 5H10 and a polyclonal rabbit antiserum against the EIII region as described (5). AZ10-75, AZ10-581 and all E-312 variants were effectively neutralized by the monoclonal antibodies and antiserum (Table 2).
Detection of a Leu→Ile mutation at residue 312 in EIII and its apparent persistence since first detection in the 2005 South Dakota isolate were major findings given the variable nature of this residue in other WNV lineages and the presumed importance of EIII in antigenicity and receptor binding activity of E protein. Although other parameters that could contribute to the selection of E-312 variants in nature remain to be explored, analysis of engineered E-312 mutants suggested that most nonsynonymous single nucleotide mutations at this site, including the Leu→Ile substitution in some AZ10 isolates, have no major effect on virulence of NY99-derived WNV in mice and were not associated with major changes in antigenicity.
The 2010 epidemic of WNV disease in the Maricopa County area was associated with co-circulation of 3 distinct WNV variants. The high mouse virulence of all strains tested suggests that signature nucleotide and amino acid changes associated with the different genotypes involved (4,10) were probably not linked to major changes in virulence for mammalian hosts, and that all 3 variants might have contributed to human disease in the 2010 outbreak. Some nonstructural protein mutations have been shown to influence virulence in avian hosts (12), and that phenotype remains to be determined for these strains or other recently identified WNV variants.
Detection of multiple sequence variants has been associated with outbreaks in the United States in as early as 2002 (13), but co-circulation of variants in relatively narrow spatial and temporal contexts, such as that observed in Maricopa County, has been a feature of recent investigations, including WNV transmission in El Paso, Texas, and Ciudad Juarez, Mexico, during 2010 (14) and during the 2012 outbreak in Dallas, Texas (15). These findings highlight the current complexity and dynamic nature of WNV transmission in the United States and suggest that co-circulation of multiple variants, with continued introduction or reintroduction of variants into disease-endemic areas, will be a major feature of future outbreaks.
Ms Plante is a doctoral student at the University of Texas Medical Branch, Galveston, TX. Her research interests include molecular biology of flaviviruses.
Acknowledgments
We thank the Maricopa County Environmental Services Vector Control Division for support and assistance during field studies that led to virus isolations; John Townsend, Kirk Smith, Dan Damian, K. Cox, and the Maricopa County Environmental Services Vector Control Division field crew for assistance; Maricopa County residents for allowing mosquito collection on their property; and James Colborn for preparing Figure 1.
This study was supported in part by funding to D.W.C.B. from the Institute for Human Infections and Immunity at the University of Texas Medical Branch. J.A.P. was supported by fellowships from the Sealy Center for Vaccine Development and the Biodefense Training Program (National Institutes of Health grant T32-AI060549).
References
- Gibney KB, Colborn J, Baty S, Bunko Patterson AM, Sylvester T, Briggs G, Modifiable risk factors for West Nile virus infection during an outbreak, Arizona, 2010. Am J Trop Med Hyg. 2012;86:895–901. DOIPubMedGoogle Scholar
- Godsey MS Jr, Burkhalter K, Young G, Delorey M, Smith K, Townsend J, Entomologic investigations during an outbreak of West Nile virus disease in Maricopa County, Arizona, 2010. Am J Trop Med Hyg. 2012;87:1125–31. DOIPubMedGoogle Scholar
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. DOIPubMedGoogle Scholar
- Añez G, Grinev A, Chancey C, Ball C, Akolkar N, Land KJ, Evolutionary dynamics of West Nile virus in the United States, 1999–2011: phylogeny, selection pressure and evolutionary time-scale analysis. PLoS Negl Trop Dis. 2013;7:e2245. DOIPubMedGoogle Scholar
- Bakonyi T, Hubalek Z, Rudolf I, Nowotny N. Novel flavivirus or new lineage of West Nile virus, central Europe. Emerg Infect Dis. 2005;11:225–31. DOIPubMedGoogle Scholar
- Li L, Barrett AD, Beasley DW. Differential expression of domain III neutralizing epitopes on the envelope proteins of West Nile virus strains. Virology. 2005;335:99–105. DOIPubMedGoogle Scholar
- McMullen AR, Albayrak H, May FJ, Davis CT, Beasley DW, Barrett AD. Molecular evolution of lineage 2 West Nile virus. J Gen Virol. 2013;94:318–25. DOIPubMedGoogle Scholar
- Papa A, Bakonyi T, Xanthopoulou K, Vazquez A, Tenorio A, Nowotny N. Genetic characterization of West Nile virus lineage 2, Greece, 2010. Emerg Infect Dis. 2011;17:920–2. DOIPubMedGoogle Scholar
- Bakonyi T, Ivanics E, Erdelyi K, Ursu K, Ferenczi E, Weissenbock H, Lineage 1 and 2 strains of encephalitic West Nile virus, central Europe. Emerg Infect Dis. 2006;12:618–23. DOIPubMedGoogle Scholar
- McMullen AR, May FJ, Li L, Guzman H, Bueno R Jr, Dennett JA, Evolution of new genotype of West Nile virus in North America. Emerg Infect Dis. 2011;17:785–93. DOIPubMedGoogle Scholar
- Zhang S, Bovshik EI, Maillard R, Gromowski GD, Volk DE, Schein CH, Role of BC loop residues in structure, function and antigenicity of the West Nile virus envelope protein receptor-binding domain III. Virology. 2010;403:85–91. DOIPubMedGoogle Scholar
- Brault AC. Changing patterns of West Nile virus transmission: altered vector competence and host susceptibility. Vet Res. 2009;40:43. DOIPubMedGoogle Scholar
- Beasley DW, Davis CT, Guzman H, Vanlandingham DL, Travassos da Rosa AP, Parsons RE, Limited evolution of West Nile virus has occurred during its southwesterly spread in the United States. Virology. 2003;309:190–5. DOIPubMedGoogle Scholar
- Mann BR, McMullen AR, Guzman H, Tesh RB, Barrett AD. Dynamic transmission of West Nile virus across the United States–Mexican border. Virology. 2013;436:75–80. DOIPubMedGoogle Scholar
- Duggal NK, D’Anton M, Xiang J, Seiferth R, Day J, Nasci R, Sequence analyses of 2012 west nile virus isolates from Texas fail to associate viral genetic factors with outbreak magnitude. Am J Trop Med Hyg. 2013;89:205–10 . DOIPubMedGoogle Scholar
Figures
Tables
Cite This Article1These authors contributed equally to this article.
Table of Contents – Volume 20, Number 2—February 2014
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
David W. C. Beasley, Department of Microbiology and Immunology, University of Texas Medical Branch, 301 University Blvd, Galveston, TX 77555-0609, USADavid W. C. Beasley, Department of Microbiology and Immunology, University of Texas Medical Branch, 301 University Blvd, Galveston, TX 77555-0609, USA
Top