Volume 30, Number 5—May 2024
Dispatch
Recurrence, Microevolution, and Spatiotemporal Dynamics of Legionella pneumophila Sequence Type 1905, Portugal, 2014–2022
Abstract
We investigated molecular evolution and spatiotemporal dynamics of atypical Legionella pneumophila serogroup 1 sequence type 1905 and determined its long-term persistence and linkage to human disease in dispersed locations, far beyond the large 2014 outbreak epicenter in Portugal. Our finding highlights the need for public health interventions to prevent further disease spread.
Legionella, the causative agent of Legionnaires’ disease (LD), is a facultative intracellular gram-negative bacterium that is ubiquitous in freshwater environments. Legionella bacteria usually thrive in natural and artificial water sources, such as cooling towers, hot water tanks, and plumbing systems. Humans acquire infection by inhaling bacteria-contaminated droplets, usually generated by water systems or devices (1). Although the primary mode of transmission is exposure to contaminated aerosolized water, not all Legionella species known to cause disease in humans are exclusively associated with water sources. For example, infection with L. longbeachae is associated with exposure to soil and compost-related products (2). Although Legionella bacteria typically do not spread from person to person, that transmission route has been reported (3,4).
Incidence of Legionella infection in a population is primarily influenced by 3 factors: individual susceptibility, environment, and type of exposure. Outbreaks often occur in healthcare settings (e.g., hospitals or long-term care facilities) because of the presence of persons at increased risk for infection and severe disease (5).
One of the world’s largest outbreaks of LD (>400 cases and 14 deaths) occurred in the Vila Franca de Xira (VFX) region of Portugal, in 2014 (6,7). The outbreak was associated with the novel sequence type (ST) 1905 of L. pneumophila subspecies fraseri serogroup 1 strain (PtVFX/2014), which probably originated from a local industrial cooling tower (6,7). In-depth genomic analyses showed that PtVFX/2014 possesses a unique and mosaic genomic backbone marked by specific evolutionary and genetic traits (3), including a recently identified novel effector with nucleotropism (8), that may affect its ability to adapt and persist in diverse environments and cause human disease. The L. pneumophila ST1905 strain was also implicated by the strongest evidence to date of person-to-person transmission of LD (3,4). To our knowledge, L. pneumophila ST1905 has not been reported in countries other than Portugal. To elucidate the molecular evolution and spatiotemporal dynamics of L. pneumophila serogroup 1 ST1905 during 2014–2022, we investigated the phylogenetic relationship of strains isolated during that period at the National Institute of Health, Portugal, in the context of the National Legionnaires’ Disease Surveillance Programme.
Figure 1
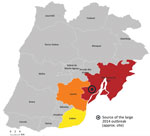
Figure 1. Geographic spread of 12 Legionella pneumophilasequence type 1905 isolates by probable municipality of exposure (clinical) or sampling (environmental), Lisbon region, Portugal, 2014–2022. Location of 2014 Legionnaires’ disease outbreak...
Figure 2

Figure 2. Core SNP-based phylogeny of whole-genome sequencing data from 12 Legionella pneumophilaisolates obtained from samples collected in 3 interconnected municipalities in the Lisbon region, Portugal. Phylogeny was generated using...
Since the large 2014 LD outbreak, the ST1905 genotype has been identified several times in Portugal. For our study, we analyzed all L. pneumophila serogroup 1 ST1905 isolates (6 clinical and 6 environmental) obtained from samples collected in 3 interconnected municipalities in the Lisbon region (Lisbon, Loures, and VFX) in the context of clinical case investigations (outbreaks or as sporadic cases) (Figures 1, 2; Appendix 1; Appendix 2). We also included 2 culture-negative clinical samples genotyped as ST1905 for analysis of the temporal and geographic distribution of this strain after 2014 (Appendices 1, 2).
Single-nucleotide polymorphism (SNP)–based diversity analysis confirmed that all ST1905 isolates were genetically similar to PtVFX/2014, differing by 3–6 SNPs. Overall, the observed microevolution across the 12 isolates was marked by 10 SNPs (9 nonsynonymous and 1 synonymous mutations), 4 insertion/deletions, and 1 small recombination event (Figure 2). The low number of SNPs during the 8-year period supports the notion that L. pneumophila evolves at a very slow rate, resulting in substantial temporal and spatial conservation, as previously reported (9,10). When compared with PtVFX/2014, all isolates presented a recombination event in an ≈2.5-kb region (contig 8, PtVFX/2014_08985-08995) belonging to the type IVA secretion system, which is associated with the survival of the bacteria in the environment (11). Of note, Lpn_PT_E259_y2021, isolated from a sporadic LD case, presented mutational events, including 2 deletions and 1 insertion, not found in any other isolates included in this study. The first deletion encompasses a 45,479-bp segment (contig 8, PtVFX/2014_09080-09280) that includes an ≈37.5-Kb genomic island containing an lvh/lvr type IVA secretion system cluster. That cluster was first identified within the L. pneumophila species in the PtVFX/2014 strain (PtVFX/2014_09115-09280), possibly because of interspecies transfer from L. oakridgensis (3). The second deletion spanned 142 bp and occurred in a CRISPR-associated intergenic region (PtVFX/2014_08935/08940). Moreover, we observed a frameshift 25-bp insertion at the PtVFX/2014_13190 locus, coding for a protein with unknown function. That locus seems to be specific for L. pneumophila subsp. fraseri and is located within a larger genomic region that contains known Dot/Icm substrates (3). Although we cannot make conclusions about a potential adaptive role of the observed mutations, it has been hypothesized that mutations found exclusively in clinical isolates, as in our study, might reflect human-specific adaptation (10). L. pneumophila can infect and replicate in human alveolar macrophages, but human-to-human transmission is assumed to be rare; thus, fixation of those mutations into L. pneumophila circulating in the human population is unlikely (10,12). Still, it has been proposed that the recent expansion of L. pneumophila in manmade water systems, together with the widespread distribution of specific clones at global scale, aligns with the potential dissemination between humans or from humans to the environment (13).
In-depth phylogenetic and microevolutionary analysis showed that ST1905 isolates did not cluster by year of isolation (Figure 2). Still, it is noteworthy that all clinical and environmental isolates associated with a particular location clustered apart, supporting persistence of the strain in that location and linkage between different events in some specific settings. Indeed, in one facility, located in the Lisbon municipality, we observed a genetic and epidemiologic correlation between the isolates collected in the outbreak investigation in 2015 and those from 2018 (Figures 1, 2; Appendix 1). In that facility, when both clinical and environmental isolates collected in the same investigation context were available, they differed by <3 SNPs, further supporting their epidemiologic linkage. Local microevolution is expected (13) and might contribute to fitness changes, such as increased tolerance to copper (14).
Three mutation events (2 SNPs and 1 recombination) were shared by all ST1905 isolates collected after the 2014 outbreak (Figure 2) We retrospectively inspected isolates from the 2014 outbreak for evidence of any of those mutations (3) and found that 1 nonsynonymous mutation (in PtVFX/2014_02480, coding for a hypothetical protein) had already been detected in 1 of the 2014 clinical strains (data not shown). That observation provides further evidence that the recurrent ST1905 detections derived from the large bacterial dispersion that occurred in 2014, probably resulting from atypical atmospheric conditions (6,7,15). It also supports the knowledge that L. pneumophila outbreaks can be caused by multiple same-strain subpopulations (present simultaneously in a source of infection and diversified over time) or even by different co-existing strains (9).
Our results strongly indicate that the atypical L. pneumophila sg1 ST1905 strain is potentially persistent in diverse and geographically dispersed environments, far beyond the epicenter of the large 2014 outbreak in Portugal (VFX region). The recurrence of isolated cases or outbreaks in other regions with susceptible populations is thus of public health concern. Moreover, the observed microevolutionary traits and the potential of genetic recombination raise additional uncertainties regarding the evolutionary landscape of the ST1905 strain and its ability to further adapt and persist in the environment and, ultimately, cause human disease. Our study highlights the need for targeted public health preparedness and control strategies, emphasizing the added value of molecular epidemiology in the surveillance and management of LD.
Dr. Manageiro is a researcher at the Department of Infectious Diseases, National Institute of Health Doutor Ricardo Jorge, Lisbon, Portugal. Her research focuses on gram-negative pathogen antibiotic resistance, with an emphasis on the roles of resistome and mobilome as linkages between human, animal, and environmental reservoirs. Dr. Borges is a researcher at the Department of Infectious Diseases, National Institute of Health Doutor Ricardo Jorge, Lisbon, Portugal. His research focuses on using genomics and transcriptomics to survey and investigate pathogens with effects on public health.
Acknowledgments
We acknowledge all health professional (in particular clinicians, pathologists, and environmental health technicians) who contribute to the National Legionnaire’s Disease Surveillances Programme.
Illumina reads generated in this study were deposited in the European Nucleotide Archive (BioProject accession no. PRJEB65289; run IDs: ERR11862821-ERR11862831) (http://www.ebi.ac.uk/ena/data/view/PRJEB65289).
We report no potential conflict of interest.
References
- Iliadi V, Staykova J, Iliadis S, Konstantinidou I, Sivykh P, Romanidou G, et al. Legionella pneumophila: the journey from the environment to the blood. J Clin Med. 2022;11:6126. DOIPubMedGoogle Scholar
- Chambers ST, Slow S, Scott-Thomas A, Murdoch DR. Legionellosis caused by non-Legionella pneumophila species, with a focus on Legionella longbeachae. Microorganisms. 2021;9:291. DOIPubMedGoogle Scholar
- Borges V, Nunes A, Sampaio DA, Vieira L, Machado J, Simões MJ, et al. Legionella pneumophila strain associated with the first evidence of person-to-person transmission of Legionnaires’ disease: a unique mosaic genetic backbone. Sci Rep. 2016;6:26261. DOIPubMedGoogle Scholar
- Correia AM, Ferreira JS, Borges V, Nunes A, Gomes B, Capucho R, et al. Probable person-to-person transmission of Legionnaires’ disease. N Engl J Med. 2016;374:497–8. DOIPubMedGoogle Scholar
- Beauté J, Plachouras D, Sandin S, Giesecke J, Sparén P. Healthcare-Associated Legionnaires’ Disease, Europe, 2008-2017. Emerg Infect Dis. 2020;26:2309–18.PubMedGoogle Scholar
- Shivaji T, Sousa Pinto C, San-Bento A, Oliveira Serra LA, Valente J, Machado J, et al. A large community outbreak of Legionnaires disease in Vila Franca de Xira, Portugal, October to November 2014. Euro Surveill. 2014;19:20991. DOIPubMedGoogle Scholar
- George F, Shivaji T, Pinto CS, Serra LAO, Valente J, Albuquerque MJ, et al. A large outbreak of Legionnaires’ disease in an industrial town in Portugal. Rev Port Saude Publica. 2016;34:199–208. DOIGoogle Scholar
- Monteiro IP, Sousa S, Borges V, Gonçalves P, Gomes JP, Mota LJ, et al. A search for novel Legionella pneumophila effector proteins reveals a strain specific nucleotropic effector. Front Cell Infect Microbiol. 2022;12:
864626 . DOIPubMedGoogle Scholar - David S, Mentasti M, Lai S, Vaghji L, Ready D, Chalker VJ, et al. Spatial structuring of a Legionella pneumophila population within the water system of a large occupational building. Microb Genom. 2018;4:
e000226 . DOIPubMedGoogle Scholar - Leenheer D, Moreno AB, Paranjape K, Murray S, Jarraud S, Ginevra C, et al. Rapid adaptations of Legionella pneumophila to the human host. Microb Genom. 2023;9:
mgen000958 . DOIPubMedGoogle Scholar - Qin T, Zhou H, Ren H, Liu W. Distribution of secretion systems in the genus Legionella and its correlation with pathogenicity. Front Microbiol. 2017;8:388. DOIPubMedGoogle Scholar
- Oliva G, Sahr T, Buchrieser C. The life cycle of L. pneumophila: cellular differentiation is linked to virulence and metabolism. Front Cell Infect Microbiol. 2018;8:3. DOIPubMedGoogle Scholar
- David S, Rusniok C, Mentasti M, Gomez-Valero L, Harris SR, Lechat P, et al. Multiple major disease-associated clones of Legionella pneumophila have emerged recently and independently. Genome Res. 2016;26:1555–64. DOIPubMedGoogle Scholar
- Bédard E, Trigui H, Liang J, Doberva M, Paranjape K, Lalancette C, et al. Local adaptation of Legionella pneumophila within a hospital hot water system increases tolerance to copper. Appl Environ Microbiol. 2021;87:e00242–21. DOIPubMedGoogle Scholar
- Russo A, Gouveia CM, Soares PMM, Cardoso RM, Mendes MT, Trigo RM. The unprecedented 2014 Legionnaires’ disease outbreak in Portugal: atmospheric driving mechanisms. Int J Biometeorol. 2018;62:1167–79. DOIPubMedGoogle Scholar
Figures
Cite This ArticleOriginal Publication Date: April 17, 2024
1These first authors contributed equally to this article.
Table of Contents – Volume 30, Number 5—May 2024
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Paulo Gonçalves, National Reference Laboratory for Legionella, Department of Infectious Diseases, National Institute of Health Doutor Ricardo Jorge (INSA), Lisbon, Portugal
Top