Volume 7, Number 5—October 2001
Synopsis
Potential Infectious Etiologies of Atherosclerosis: A Multifactorial Perspective
Cite This Article
Citation for Media
Abstract
Coronary heart disease (CHD) contributes substantially to illness and death worldwide. Experimental studies demonstrate that infection can stimulate atherogenic processes. This review presents a spectrum of data regarding the link between CHD and infection. In addition, the need for improved diagnostic tools, the significance of multiple pathogens, and potential intervention strategies are discussed.
Cardiovascular disease (CVD) from all causes accounts for 29% of deaths worldwide and ranks second only to infectious and parasitic diseases (1). Deaths from CVD are often premature, and millions of nonfatal events result in disability. Atherosclerosis, a major component of CVD, has properly been considered a public health problem of industrialized countries, accounting for an estimated one third of deaths overall (1). In the United States alone, atherosclerosis reportedly affects one in four persons, causing approximately 42% of all deaths. Approximately half of these are due to atherosclerotic coronary heart disease (CHD) (2-4). Atherosclerotic CVD now threatens developing countries as well, perhaps reflecting greater exposure to certain risk factors with rising standards of living (5). In India, for example, higher socioeconomic status correlates with increases in risk factors for and rates of CVD (5). Thus, the need for public health measures to limit its impact is expanding worldwide.
Many persons with atherosclerosis, however, lack identifiable traditional risk factors. The well-recognized influences of smoking, diet and exercise, hypercholesterolemia, hypertension, diabetes, and some genetic factors account for much less than 100% of disease. Despite continued reductions in the prevalence of modifiable risk factors, the steady, two-decade decrease in cardiovascular mortality rates in western industrialized countries has reached a plateau (2-4). In contrast with India, death rates for chronic (including atherosclerotic) and infectious diseases in the United States are inversely related to education and income (4-6). These observations fuel renewed interest in a link between atherosclerotic CVD and as-yet poorly defined environmental exposures, including infectious agents. If epidemiologic and laboratory evidence eventually supports the association, atherosclerosis could emerge as another noncommunicable chronic condition related to infection.
How strong is the link? This review examines several candidate infectious agents, referring to reports and reviews by investigators summarizing their own work and a body of research. It discusses the potential roles of less well-established factors, interactions between infection and traditional risk factors, and unanswered questions on etiology, pathogenesis, and appropriate interventions. Developing an appropriate research agenda and public health response requires clarification of these issues. The following discussion provides a foundation for future studies.
Atherogenic processes resemble many aspects of chronic inflammation (7), a response that may be promoted by microorganisms (8-10). Accordingly, reviews have revisited the venerable hypothesis of an infectious etiology (8,9,11). Experimental animal studies have shown that bacterial and viral agents could contribute to atherogenesis. Both Chlamydia pneumoniae and cytomegalovirus (CMV), for example, are widely distributed, can infect blood vessel wall cells, and exhibit persistence, latency, and recurrence of infection. However, the potential mechanisms of infection-induced atherosclerosis remain speculative.
Figure 1
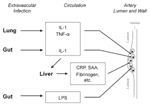
Figure 1. . "Echo" hypothesis: activation of atheroma-associated cells by bacterial products and cytokines released in response to extravascular infection. a. Extravascular infection stimulates production of inflammatory cytokines, e.g. interleukin-1 (IL-1) and tumor...
The earliest lesions of atherogenesis consist of arterial intimal accumulations of foam cells (primarily lipid-laden macrophages) and T lymphocytes intermixed with smooth muscle cells (7). Infection could indirectly influence this process without infiltrating the artery wall (Figure 1). Host defenses to extravascular infections usually elicit proinflammatory cytokines and stimulate increased expression of cellular adhesion molecules, enhancing leukocyte adhesion. These cytokines could elicit a second wave or "echo" from inflammatory cells already at sites of atherogenesis, such as arterial wall cells or macrophages (12). Circulating microbial products such as endotoxin can also produce an echo. Similarly, cytokines induced by extravascular infection (specifically interleukin-6) characteristically elicit hepatic synthesis of acute-phase reactants, some of which might promote atheromata complicated by thrombosis. Accordingly, levels of the acute-phase reactant fibrinogen correlate prospectively with risk for coronary events, and plasminogen activator inhibitor can promote clot stability by interfering with fibrinolysis (7-10). Still, direct infection of the arterial wall could promote evolution of atherosclerotic lesions or precipitate acute cardiovascular events. These potential effects are discussed below.
Chlamydia pneumoniae Association
Lines of evidence associating C. pneumoniae with atherosclerosis include seroepidemiologic studies, direct detection of bacterial components in atherosclerotic lesions, occasional isolation of viable organisms from coronary and carotid atheromatous tissue, and in vitro and animal experiments (reviewed in 8,9,13,14). Most cross-sectional and prospective studies have correlated seroprevalence with myocardial infarction, chronic CHD, or stroke (8,13,15). However, the diagnostic criteria used, cut-off titers to define seropositivity, adjustment for confounders (e.g., smoking), and sample sizes have varied notably. Indeed, as the interrelationships of smoking, C. pneumoniae infection, socioeconomic status, and atherosclerotic CVD are further investigated, epidemiologists continue to discuss which data adjustments are most appropriate (16-19). Interpretation of these studies is complicated by a lack of standardized serologic methods and by interlaboratory variations and poor reproducibility in microimmunofluorescence test results (20-22). Temporal variations in antibody and antigen titers and the high prevalence of infection--more than 50% of adults have been infected at least once--also complicate interpretation (13). Still, more than 38 studies have reported a positive association between antibodies to C. pneumoniae and atherosclerotic disease. Only four studies did not confirm this association, including two recent, large, well-controlled investigations in which anti-C. pneumoniae immunoglobulin (Ig) G titers did not correlate prospectively with risk for coronary events in men or women (18,23-25). Additionally, the prospective Atherosclerosis Risk in Communities Study correlated IgG titers >1:64 with incident CHD, but the relationship did not persist after the data were adjusted for several cardiovascular risk factors (26).
The strongest evidence associating C. pneumoniae with atherosclerotic CVD has been detection of bacterial components in atherosclerotic lesions. C. pneumoniae appears to have a tropism for atheromata. It is rarely found in normal arteries or infectious and noninfectious granulomas and is documented more frequently in atheromata than in lung or other tissue from the same patient (13). Over 30 peer-reviewed publications from investigators worldwide, using different diagnostic methods, have localized C. pneumoniae antigen, DNA, or both in atheromata; three reports did not find such components (8,27,28). Although detection rates have varied depending on the diagnostic methods used, the cumulative evidence supports existence of the organism in many lesions (8,27). These histologic findings, however, do not establish a causal role for C. pneumoniae in atherogenesis. Despite improved yields, culture of C. pneumoniae from atheromata remains difficult (8,13,29). Moreover, while the presence of a viable microbe or its components at a site suggests that it may initiate or exacerbate disease, it does not prove pathogenesis.
Detection of C. pneumoniae antigens or DNA in intimal thickening and fatty streaks of young adults and Alaskan Natives (the latter group at low risk for coronary atherosclerosis) supports an early microbial role in pathogenesis (30). Postmortem, the Alaskan retrospective study also positively correlated prior systemic infection with evidence of C. pneumoniae in atherosclerotic lesions; studies from Seattle reported a slightly higher detection rate in late-stage lesions (13). Recently, investigators reported a statistically significant relationship between the presence of C. pneumoniae and the severity of human coronary atherosclerosis (31). Although others observed no correlation between C. pneumoniae and disease severity in the same coronary artery, evidence of the bacterium was found in 80% of patients' arteries examined (32).
Detection of C. pneumoniae in plaques has not correlated well with serology (8,33), so investigators have attempted to predict endovascular infection through polymerase chain reaction (PCR) detection of microbial DNA in peripheral blood monocytes. The prevalence of C. pneumoniae DNA in these mononuclear cells has varied between studies (perhaps due to differences in both assay sensitivity and extraction procedures), but was 59% in coronary angiography patients compared with 44% in blood donors in one series; the rate appears to increase with age (33,34). Several but not all investigators have now correlated PCR-positive mononuclear cells with clinical CHD (34,35) or the detection of C. pneumoniae nucleic acid in atherosclerotic aortic tissue (36).
Figure 2
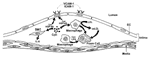
Figure 2. . Possible direct effects of Chlamydia pneumoniae (Cpn) on atheromata. Cpn infection augments endothelial cell production of inflammatory cytokines and expression of adhesion molecules, e.g., vascular cell adhesion molecule (VCAM)-1, enhancing...
In vitro studies support hypotheses that C. pneumoniae might directly promote atherosclerosis (Figure 2). Infection of human endothelial cells augments their production of inflammatory cytokines and modulates expression of adhesion molecules, enhancing recruitment of inflammatory leukocytes to the vessel wall (8,37,38). Chlamydial endotoxins, much less virulent than those of enterobacteriaceae (e.g., Escherichia coli), can promote macrophage foam cell formation in vitro (8,39). Furthermore, C. pneumoniae heat shock protein (HSP-60) can activate both macrophage tumor necrosis factor-alpha and expression of matrix-degrading proteinases that may weaken atherosclerotic plaques, rendering them susceptible to rupture and hence thrombosis. HSP-60 can induce proinflammatory activities in macrophages and vascular cells (e.g., endothelium, smooth muscle cells) and promote oxidation of lipoproteins by macrophages (10,40).
Animal experiments have also explored the link between C. pneumoniae and atherosclerosis. Intranasal or intratracheal C. pneumoniae inoculation of New Zealand white rabbits fed a normal diet produced inflammatory changes of the aorta (41-43). Although one group did not observe foam cells in inflamed sites, another has reported foam cells in lesions resembling early atherosclerosis. Macrolide antibiotic therapy appeared to dampen the additive effects of hypercholesterolemia and serial C. pneumoniae infection (9,41,43); macrolide antibiotics with antichlamydial activity administered early in infection could prevent changes, but delayed treatment had little or no effect (41). These observations, along with diminished effects on cholesterol-induced lesions (in the absence of infection), suggest antimicrobial mechanisms of action.
Inflammatory changes without foam cell lesions and typical atheromata have also been observed in C. pneumoniae-infected mice fed a normal chow diet (13,44). However, C. pneumoniae infection does potentiate the atherogenic effects of hypercholesterolemia in three mouse models. Intranasal inoculation of apolipoprotein E-deficient mice fed a regular chow diet accelerated aortic arch plaque development (45); similar changes occurred in low-density lipid receptor-deficient mice on a high-cholesterol regime but not those on a regular diet (46). In C57BL/6J mice fed a high-fat, high-cholesterol diet, C. pneumoniae infection accelerated the development of atherosclerotic lesions in the aortic sinus (44,47).
Although experimental animal lesions do not mimic human atherogenesis exactly, similarities have prompted human intervention studies directed against C. pneumoniae infection. Three prospective therapeutic trials have been reported, but all had insufficient statistical power to resolve the question (reviewed in 23,48). One described a fivefold decrease in cardiovascular events among seropositive men treated with short courses of azithromycin after myocardial infarction (MI) (49). In a double-blind study of unstable angina or non-Q wave infarction, patients treated for 30 days with roxithromycin had a decrease in cardiovascular events that waned by 6 months (50). Additionally, when 302 MI survivors with C. pneumoniae antibody titers >1:16 were treated for 3 months with azithromycin, investigators observed no significant reduction in recurrent events at 6 and 24 months, although a gradual decline in serologic (inflammatory) markers of cardiovascular risk was initially seen (51,52). Three disparate retrospective, multivariate analyses of antibiotic use and CHD risk, using different methods and populations, also reported both positive and negative correlations between past antibiotic prescriptions and CHD or MI risk (48). At this time, any specific effects of treatment remain unclear (23).
Cytomegalovirus Links
Almost two decades ago, several investigators suggested a role for herpesviruses in CVD. Lesions resembling human atheromata developed in pathogen-free chickens infected with Marek disease virus, an avian herpesvirus; immunization with turkey herpesvirus was protective. In vitro, infected arterial smooth muscle cells accumulated cholesterol. Since then, seroepidemiologic, histopathologic, in vitro, and animal studies have investigated possible links between human atherosclerosis and human herpesviruses, primarily CMV (reviewed in 14,15,53-55).
Studies have linked CMV to three arterial diseases: primary atherosclerosis, post-angioplasty restenosis, and post-transplantation arteriosclerosis. For each, seroepidemiology has relied on single measures of viral IgG antibodies, which only indicate previous exposure. Similar to C. pneumoniae, the worldwide ubiquity of lifelong, latent CMV infections, temporal variations in antibody or antigen titers, and viral reactivation could mask or falsely highlight causality. These factors may explain inconsistencies in cross-sectional, retrospective, and prospective studies attempting to correlate seroprevalence or higher IgG titer with primary atherosclerosis (10,14,15,24,56-58). Differing disease classifications (based on histopathology of vascular surgery, general autopsy or atherectomy specimens, angiography, noninvasive carotid imaging, or clinical history) and epidemiologic methods (small sample sizes, diminished statistical significance after data adjustment, nonuniform consideration of other risk factors) also limit interpretation (14,53,55). Results are conflicting even in prospective, nested case-control investigations focused on but not limited to male Caucasians. The Atherosclerosis Risk in Communities Study correlated preexisting high anti-CMV titers and traditional risk factors with carotid atherosclerosis, incident MI or CHD death (relative risk 1.76; 95% confidence interval 1.00-3.11) during a 5-year follow-up period (57). In contrast, the 12-year Physician's Health Study found no association between antibody prevalence and subsequent first MI or thromboembolic stroke, or elevated C-reactive protein (56), while the Cardiovascular Health Study correlated antibodies to herpes simplex virus type 1 but not CMV or C. pneumoniae with incident events in the elderly (59).
In post-angioplasty restenosis, preexisting and high-titer IgG CMV antibodies predicted restenosis (43% vs. 8%) independently of CVD risk factors, but the sample size was small and no follow-up titer analysis was included (53,60,61). Cross-sectional and prospective studies also link CMV to post-transplant arteriosclerosis (14,62-64), in which a subset of immunosuppressed recipients manifests accelerated disease in the first year.
Evidence of CMV in these disease entities, however, does not prove causality. Different techniques have detected viral antigens and nucleic acids in 0% to 94% of plaques (mean 40%-50%, usually localized to the periphery of advanced plaques), but also in most of the uninvolved arteries from the same patients and >50% of random arterial specimens (14,54,65). A recent PCR analysis of coronary arteries and venous bypass graft occlusions found no CMV in specimens but did detect C. pneumoniae (66). Reports of viral nucleic acids in post-transplantation arteriosclerotic lesions, using different techniques, are not uniform (62,63,67,68).
In vitro observations support a potential etiologic role for CMV in atherosclerotic CVD. Indirectly, virally provoked paracrine expression of chemokines and growth factors might stimulate migration of smooth muscle cells from the adventitia and media into the intima, along with smooth muscle cell proliferation, both important components of atherogenesis and restenosis (54). Direct infection of arterial smooth muscle cells appears to augment expression of the CMV chemokine receptor US28 (69). Investigators have also reported inhibited apoptosis and augmented cellular proliferation when the virally directed protein IE2-84 binds or inhibits transcriptional activity of p53 (54,70). Direct CMV infection may also enhance uptake of oxidized low-density lipoprotein by macrophages and smooth muscle cells, promoting foam cell development; through its immediate-early gene products, CMV may increase uptake of modified low-density lipoprotein and stimulate type A scavenger receptor gene expression by smooth muscle cells (54).
CMV animal experiments also do not duplicate human disease but can provide insight on causality. While CMV infection of rats several months preceding or during balloon injury to the carotid artery augmented neointimal thickening (54,71), infection 14 days after injury did not (72). In ApoE-deficient mice, systemic infection was reported to increase atherosclerotic lesion size (54); local infection accentuated subendothelial inflammation and coronary arteriosclerosis in rat cardiac and aortic allografts, apparently reduced by ganciclovir (DHPG) treatment (55,73). Thus, CMV remains potentially linked to the promotion of atherosclerotic CVD.
Oral Pathogens
During the past decade, several reports have suggested a relationship between chronic oral infections (e.g., periodontitis) and cardiovascular disease (74). Among the Pima Indians, periodontitis emerged as an independent risk factor for atherosclerotic events (74). Other studies described an association between tooth loss related to periodontal disease and CVD (75), while persons with diabetes have a higher incidence of infections (including oral) and atherosclerotic CVD. Pathogens potentially associated with CVD include Porphyromonas gingivalis, Bacteroides forsythus, Campylobacter rectus, Fusobacterium nucleatum, Treponema spp., and Prevotella species. At least two, P. gingivalis (42%) and Streptococcus sanguis (12%), were identified in the periphery of atherosclerotic plaques by immunologic stain (65). Another group detected >2 periodontal pathogens by PCR for 16SrDNA in 13 of 22 endarterectomy specimens (76).
Researchers have hypothesized that certain persons with a hyperinflammatory phenotype are at increased risk for both periodontitis and atherosclerotic CVD, with dietary and other factors possibly altering disease progression (reviewed in 74). Like C. pneumoniae, oral bacteria might affect atherosclerosis through direct invasion of vascular endothelial cells or indirectly through products that stimulate proinflammatory and prothrombotic functions of vascular cells. For example, P. gingivalis and S. sanguis both express a platelet-aggregation factor (74). However, the true links between oral bacteria and atherosclerotis are still to be determined, requiring further laboratory and carefully adjusted clinical studies.
Helicobacter pylori, Herpes Simplex Virus (HSV), and Other Pathogens
The literature linking H. pylori, HSV-1, and HSV-2 to atherogenesis is less extensive than for C. pneumoniae or CMV (9,14,15,25,53,59). H. pylori has not been detected in human atheromata, and available seroepidemiologic data conflict, weakening the proposed etiologic association (9,14,15,25). Social factors linked to CVD and H. pylori infection, including socioeconomic status, may confound these reported associations, and data were not adjusted appropriately in most studies (15,77). Nonetheless, an indirect role for H. pylori in the pathogenesis of atherosclerosis has not been disproved.
Unlike CMV, HSV-1 has a predilection for epithelial and neuronal tissues, not mononuclear cells. Seroepidemiologic and histopathologic study results vary widely (53,55-57,59). For example, the prospective, nested Atherosclerosis Risk in Communities Study found no association between incident CHD and preexisting HSV-1 antibodies (57). Although consistent with most other analyses (25,53,56), this contrasts with positive HSV-1 results from the elderly Cardiovascular Health Study cohort (59).
In summary, laboratory investigations suggest but do not conclude that herpesviridae could directly and indirectly induce several endothelial cell responses involved in atherothrombosis (78). The long-term effects of commonly silent or chronic herpesvirus or other persistent infections render plausible a potential role in atherosclerotic CVD.
Chronic diseases, including atherosclerosis, have complex causal mechanisms. Accepting the infection hypothesis for atherosclerotic CVD depends on several key issues, including those discussed below.
Concept of Multiple Pathogens
Long-term exposure to proinflammatory, toxic, or transforming microbes and their products is a possible mechanism for infection-related atherosclerotic CVD, as is exposure to the aggregate effects of coinfections, detected or undetected. Screening for C. pneumoniae, CMV, and HSV-1, one group of investigators detected 2-3 microbes in 32% of atherosclerotic lesions, mostly in plaque shoulders and inflammatory infiltrates; 71% of the organisms identified were C. pneumoniae. Expanding the analysis, this group reported a range of 1-4 microbes in atheromata, including the oral pathogens P. gingivalis and S. sanguis (65). These findings do not confirm pathogenesis, but do highlight the possibility of synergy between organisms or a cumulative infectious effect in atherogenesis. As emphasized at a 1998 National Institutes of Health workshop, research on associations between infection and atherosclerotic CVD must consider the coinfluence of multiple infectious agents (79).
Investigators are testing the hypothesis of dose-response relationships between atherosclerotic CVD and the number of microbes to which a person has been exposed (54). For example, a recent prospective cohort related expression of antibodies against several pathogens to MI or death in patients with angiographically documented coronary artery disease (80). Perhaps cardiovascular risk increases with cumulative or earlier exposure to more or specific microbes (potentially atherogenic). C. pneumoniae infection of cells in vitro may activate a reporter gene controlled by the CMV major immediate-early promoter (81). In-vivo co-infection might reactivate latent CMV infection, increase the activity of persistent virus, or both.
Integration of Infection into a Multifactorial View of CHD Risk
Infection as a potential CVD risk factor requires consideration in context. Although interventions targeting traditional risk factors have often reduced clinical disease, considerable impact persists. Perhaps microbes enhance the harmful effects of traditional causes; for example, foam cells in dyslipedemia-induced lesions might respond to bacterial inflammatory products. Genetic and gender predispositions could determine an association between periodontitis and atherosclerosis (74). Physical activity, smoking, and lipids could modulate immune status and thus individual susceptibility to acute, reactivated, or persistent infections that are potentially important in atherogenesis (4,22,79).
Gender and nutrition can each influence immune responses to infection and atherothrombotic disease. Supporting sex determination of atherosclerotic CVD, the Pathological Determinants of Atherosclerosis in Youth study found more extensive fatty streaks in the right coronary arteries and thoracic aortas of men, but greater abdominal aortic disease in women (82). Among primates, which have diet-induced atheromata resembling those of humans, premenopausal female monkeys develop less atherosclerosis than males (83); females with low levels of social stress developed the least disease, stressed males the most. Could gender affect susceptibility to or immune response following atherogenesis-inducing infection? Reports that sex steroid hormones (e.g., estrogen) can modulate endothelial cell production of microbial-induced proinflammatory factors such as cytokines, endothelin-1, angiotensin II, and nitric oxide suggest a single common pathway (84).
Nutritional effects on atherosclerosis also extend beyond hypercholesterolemia, hypertriglyceridemia, and obesity. Years of rigorous investigation have linked antioxidant consumption to coronary artery disease, and the antioxidant vitamin E suppresses viral infection (85). However, results of antioxidant vitamin supplementation on reduction of atherosclerotic risk have generally proven disappointing (85,86). Nevertheless, if nutrition affects inflammatory response to potentially atherogenic infections, adjunct therapies could exploit nutritional supplementation in high-risk populations.
Methods Limit Conclusions
Clarification of the above issues requires improved analytic tools. Current reagents and methods are not sufficient to prove or refute a potential infectious etiology in atherosclerotic CVD. Public health officials, laboratory investigators, and clinicians participating in two 1988 workshops emphasized that deficiencies in these tools weaken conclusions (22,79). As an example, culturing C. pneumoniae from clinical specimens is difficult, and serology has relied on tedious, reagent- and reader-dependent microimmunofluorescence; immunocytochemistry and PCR substrates and techniques vary between laboratories. Temporal fluctuations in bacteremia or viremia can alter PCR analysis of peripheral blood monocytes, and no assay yet differentiates the presence of any microbe from its role in pathogenesis. Study comparability and reproducibility will require standardized, sensitive, and specific reagents (20-22).
Adding to the ambiguity, human studies have focused on late-stage atherothrombotic disease, while animal studies have concentrated on earlier stages of atherogenesis (i.e., initiation). These issues are critical for translating experimental evidence to human disease because childhood exposures to ubiquitous infections are common, frequencies of reexposure are unknown, and the age at which microbes might influence atherosclerosis is uncertain. Additionally, available analytic tools do not characterize the roles of past, active, persistent latent, or recurrent infection. Recommendations from a recent international workshop sponsored by the Centers for Disease Control and Prevention and the Laboratory Centre for Disease Control (Canada) seek to improve the reproducibility and comparability of C. pneumoniae investigations (20,21). In the future, more sensitive and specific assays, including microarray technologies (e.g., pathogen-specific identification chip) may also enhance specimen analysis (21).
Antimicrobial Therapy: Potential Benefits and Risks
If evidence substantiates a link between infection and atherosclerotic CVD, targeted antimicrobial therapy might mitigate atherosclerosis in persons at risk; controlling infections might decrease the impact of disease. Inappropriate antimicrobial therapy, however, could accelerate development of resistance in both associated and nontargeted organisms, without changing disease outcomes. Studies linking level of antibiotic use and prevalence of resistant bacteria in hospitals and communities justify these concerns; trends toward more frequent erythromycin- and penicillin-resistant pneumococci with higher antibiotic consumption reversed when macrolide use was reduced (87-89).
Although clinical antibiotic resistance in C. pneumoniae has not been described, certain characteristics--its sometimes asymptomatic, chronic, or recurrent nature--may favor this development (87). Indeed, laboratories have described antibiotic-resistant strains of C. trachomatis, a closely related organism. Antimicrobial delivery to intracellular C. pneumoniae is not well described, efficacy during different stages of the life cycle (e.g., infectious elementary bodies, replicative reticulate bodies) is unclear (8,87), and antibody titers do not reliably track infection (87). Whether antibiotics can eradicate or suppress C. pneumoniae replication, particularly in the persistent or latent state, remains conjectural. Clinically, azithromycin resistance in any Chlamydia spp. has not been extensively evaluated; however, a transient, rebound increase in resistant pneumococci has been observed following single-dose therapy for trachoma (90), and increased carriage of macrolide-resistant microbes has accompanied macrolide therapy for otitis media (91).
Widespread or long-term antibiotic use in atherosclerotic CVD could adversely affect beneficial commensals that provide nutrients and inhibit pathogen overcolonization, as well as increase antimicrobial resistance in unrelated microbes. Similar issues may emerge with antiviral therapies.
Vaccines
If antibiotics or antiviral agents do not limit the pathogenesis of acute, latent, or chronic active infections, vaccines are an alternative approach to prevention or treatment. For example, anti-Chlamydia vaccines have been sought for decades. Recent advances in immunologic techniques and description of the complete C. pneumoniae genomic sequence permit identification of antigens that may elicit protective immunity (92,93). However, poor understanding of host defense mechanisms still impedes the development and application of C. pneumoniae and other microbial vaccines. The essential components of humoral and cell-mediated immune responses associated with protection must be identified before prophylactic vaccine trials against any pathogen are implemented. To minimize the risk of stimulating autoimmunity, concerns for cross-reactivity during immunization must be addressed as vaccines against C. pneumoniae and other pathogens are pursued (94,95).
The current interest in infectious causes of atherosclerosis revives a venerable hypothesis. If true, the global prevention and intervention opportunities could be substantial (demonstrated with H. pylori in peptic ulcer disease). However, the seroepidemiologic data that triggered this renaissance lack robustness because of confounding factors (e.g., socioeconomic status, smoking) and publication bias towards more positive results. Indeed, more rigorously controlled observational and prospective studies appear to weaken the seroepidemiologic links between infection and atherosclerosis. Nonetheless, studies of the vascular biology of infection clearly demonstrate that agents such as C. pneumoniae and CMV can elicit potentially pathogenic functions of vascular wall cells and leukocytes in atheromata, supporting the hypothesis that such infections may potentiate atherosclerotic CVD. Results will soon be available from prospective clinical trials that critically examine whether macrolide antibiotic therapy reduces recurrent coronary events or modifies CHD. However, these trials will not determine eradication or suppression of one or more microbes, optimal timing of intervention, and duration of benefits, and thus fall short of providing convincing evidence for a causal relationship between infectious agents and atherosclerosis. These trials will neither define the portion of atherosclerotic CVD caused by infection nor indicate benefits or risks of vaccine therapy.
Routine use of antibiotics to prevent atherosclerotic events should await evidence from sufficiently powered and well-designed clinical trials. Premature, inappropriate use has potential adverse effects. Clearly, research on potential infection-atherosclerosis links must continue, complemented by a public health approach. Participants in recent workshops and symposia have evaluated gaps in the evidence and outlined ways to fill them (22,23,79,96,97). All agree on the need for more specific, sensitive, and standardized reagents and assays (20-22).
Research must also address interactions between infection, traditional risk factors, and other determinants of host susceptibility, such as gender and nutrition, in multifactorial atherosclerotic CVD (7,22,79,96). It will be critical to determine the age at which pathogenesis begins and to identify the attributable fraction of preventable or reversible disease and the subpopulations at risk. Such data might eventually warrant adoption of infection control strategies--minimized exposure, vaccination, and antimicrobial therapy--aimed at reducing the impact of symptomatic cardiovascular disease.
Dr. O'Connor is Assistant to the Director of the National Center for Infectious Diseases, Centers for Disease Control and Prevention. Her research interests include identification, characterization, and prevention of infectious diseases that may trigger or determine chronic illness and disability.
Acknowledgment
We thank J.M. Hughes, D. Mangan, T. Quinn, S. Skarlatos, and G. Vercellotti for review and suggestions and K. Williams for editorial assistance.
References
- World Health Organization. The World Health Report 1997. Geneva: World Health Organization; 1997.
- Centers for Disease Control and Prevention. Achievements in public health, 1900-1999: Decline in deaths from heart disease and stroke--United States, 1900-1999. Hyattsville (MD): U.S. Dept. of Health and Human Services, Centers for Disease Control and Prevention; 1999. p.649-56.
- American Heart Association. 2001 heart and stroke statistical update. Dallas (TX): American Heart Association;2000.
- Centers for Disease Control and Prevention. Chronic Disease Notes and Reports. 1997;10:2–15.
- Reddy S, Yusuf S. Emerging epidemic of cardiovascular disease in developing countries. Circulation. 1998;97:596–601.PubMedGoogle Scholar
- Centers for Disease Control and Prevention. Health, United States, 1998. Hyattsville (MD): U.S. Dept. of Health and Human Services, Centers for Disease Control and Prevention; 1998.
- Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–26. DOIPubMedGoogle Scholar
- Gaydos CA, Quinn TC. The role of Chlamydia pneumoniae in cardiovascular disease. Adv Intern Med. 2000;45:139–73.PubMedGoogle Scholar
- Muhlestein JB. Chronic infection and coronary artery disease. Med Clin North Am. 2000;84:123–48. DOIPubMedGoogle Scholar
- Kol A, Libby P. Molecular mediators of arterial inflammation: A role for microbial products? Am Heart J. 1999;138:450–2. DOIPubMedGoogle Scholar
- Nieto FJ. Infections and atherosclerosis: new clues from an old hypothesis? Am J Epidemiol. 1998;148:937–58.PubMedGoogle Scholar
- Clinton SK, Fleet JC, Loppnow H, Salomon RN, Clark BD, Cannon JG, Interleukin-1 gene expression in rabbit vascular tissue in vivo. Am J Pathol. 1991;138:1005–14.PubMedGoogle Scholar
- Campbell LA, Kuo C-C, Grayston JT. Chlamydia pneumoniae and cardiovascular disease. Emerg Infect Dis. 1998;4:571–9. DOIPubMedGoogle Scholar
- Mattila KJ, Valtonen VV, Nieminen MS, Asikainen S. Role of infection as a risk factor for atherosclerosis, myocardial infarction and stroke. Clin Infect Dis. 1998;26:719–34. DOIPubMedGoogle Scholar
- Danesh J, Collins R, Peto R. Chronic infections and coronary heart disease: is there a link? Lancet. 1997;350:430–6. DOIPubMedGoogle Scholar
- Karvonen M, Tuomilehto J, Pitkäniemi J, Naukkarinen A, Saikku P. Importance of smoking for Chlamydia pneumoniae seropositivity. Int J Epidemiol. 1994;23:1315–21. DOIPubMedGoogle Scholar
- O'Neill C, Murray LJ, Long GML, O'Reilly DPJ, Evans AE, Bamford KB. Epidemiology of Chlamydia pneumoniae infection in a randomly selected population in a developed country. Epidemiol Infect. 1999;122:111–6. DOIPubMedGoogle Scholar
- Danesh J, Whincup P, Walker M, Lennon L, Thomson A, Appleby P, Chlamydia pneumoniae IgG titres and coronary heart disease: prospective study and meta-analysis. BMJ. 2000;321:208–12. DOIPubMedGoogle Scholar
- West R. Commentary: adjustment for potential confounders may have been taken too far. BMJ. 2000;321:213.PubMedGoogle Scholar
- Peeling RW, Wang S-P, Grayston JT, Blasi F, Boman J, Clad A, Chlamydia pneumoniae serology: interlaboratory variation in microimmunofluorescence assay results. J Infect Dis. 2000;181(Suppl 3):426–9. DOIPubMedGoogle Scholar
- Dowell SF, Peeling RW, Boman J, Carlone GM, Fields BS, Guarner J, Standardizing Chlamydia pneumoniae assays: Recommendations from the Centers for Disease Control and Prevention (USA) and the Laboratory Centre for Disease Control (Canada). Clin Infect Dis. 2001. In press. DOIPubMedGoogle Scholar
- O'Connor S. Workshop on the Potential Role of Infectious Agents in Cardiovascular Disease and Atherosclerosis [News and Notes]. Emerg Infect Dis. 1999;5:186–7. DOIGoogle Scholar
- Grayston JT. Background and current knowledge of Chlamydia pneumoniae and atherosclerosis. J Infect Dis. 2000;181(Suppl 3):402–10. DOIPubMedGoogle Scholar
- Ridker PM, Kundsin RB, Stampfer MJ, Poulin S, Hennekens CH. Prospective study of Chlamydia pneumoniae IgG seropositivity and risks of future myocardial infarction. Circulation. 1999;99:1161–4.PubMedGoogle Scholar
- Ridker PM, Hennekens CH, Buring JE, Kundsin R, Shih J. Baseline IgG antibody titers to Chlamydia pneumoniae, Helicobacter pylori, herpes simplex virus, and cytomegalovirus and the risk for cardiovascular disease in women. Ann Intern Med. 1999;131:573–7.PubMedGoogle Scholar
- Nieto FJ, Folsom AR, Sorlie PD, Grayston JT, Wang S-P, Chambless LE, Chlamydia pneumoniae infection and incident coronary heart disease: The Atherosclerosis Risk in Communities Study. Am J Epidemiol. 1999;150:149–56.PubMedGoogle Scholar
- Kuo C-C, Campbell LA. Detection of Chlamydia pneumoniae in arterial tissues. J Infect Dis. 2000;181(Suppl 3):432–4. DOIPubMedGoogle Scholar
- Taylor-Robinson D, Thomas BJ. Chlamydia pneumoniae in atherosclerotic tissue. J Infect Dis. 2000;181(Suppl 3):437–40. DOIPubMedGoogle Scholar
- Maass M, Bartels C, Kruger E, Engel PM, Dalhoff K. Endovascular presence of Chlamydia pneumoniae DNA is a generalized phenomenon in atherosclerotic vascular disease. Atherosclerosis. 1998;140(Suppl 1):S25–30. DOIPubMedGoogle Scholar
- Davidson M, Kuo C-C, Middaugh JP, Campbell LA, Wang SP, Newman WP III, Confirmed previous infection with Chlamydia pneumoniae (TWAR) and its presence in early coronary atheroschlerosis. Circulation. 1998;98:628–33.PubMedGoogle Scholar
- Erickson K, Saldeen TG, Lindquist O, Pahlson C, Mehta JL. Relationship of C. pneumoniae infection to severity of human coronary atherosclerosis. Circulation. 2000;101:2568–70.PubMedGoogle Scholar
- Thomas M, Wong Y, Thomas D, Ajaz M, Tsang V, Gallagher PJ, Relation between detection of Chlamydia pneumoniae DNA in human coronary arteries at postmortem examination and histological severity (Stary grading) of associated atherosclerotic plaque. Circulation. 1999;99:2733–6.PubMedGoogle Scholar
- Boman J, Söderberg S, Forsberg J, Birgander LS, Allard A, Persson K, High prevalence of Chlamydia pneumoniae DNA in peripheral blood mononuclear cells in patients with cardiovascular disease and in middle-aged blood donors. J Infect Dis. 1998;178:274–7. DOIPubMedGoogle Scholar
- Boman J, Gaydos C. Polymerase chain reaction detection of Chlamydia pneumoniae in circulating white blood cells. J Infect Dis. 2000;181(Suppl 3):452–4. DOIPubMedGoogle Scholar
- Wong Y-K, Dawkins KD, Ward ME. Circulating Chlamydia pneumoniae DNA as a predictor of coronary artery disease. J Am Coll Cardiol. 1999;34:1435–9. DOIPubMedGoogle Scholar
- Blasi F, Boman J, Esposito G, Melissano G, Chiesa R, Consentini R, Chlamydia pneumoniae DNA detection in peripheral blood mononuclear cells is predictive of vascular infection. J Infect Dis. 1999;180:2074–6. DOIPubMedGoogle Scholar
- Molestina RE, Miller RD, Ramirez JA, Summersgill JT. Infection of human endothelial cells with Chlamydia pneumoniae stimulates transendothelial migration of neutrophils and monocytes. Infect Immun. 1999;67:1323–30.PubMedGoogle Scholar
- Gaydos CA, Summersgill JT, Sahney NN, Ramirez JA, Quinn TC. Replication of Chlamydia pneumoniae in vitro in human macrophages, endothelial cells, and aortic artery smooth muscle cells. Infect Immun. 1996;64:1614–20.PubMedGoogle Scholar
- Kalayoglu MV, Hoerneman B, LaVerda D, Morrison SG, Morrison RP, Byrne GI. Cellular oxidation of low-density lipoprotein by Chlamydia pneumoniae. J Infect Dis. 1999;180:780–90. DOIPubMedGoogle Scholar
- Kol A, Bourcier T, Lichtman AH, Libby P. Chlamydial and human heat shock protein 60s activate human vascular endothelium, smooth muscle cells, and macrophages. J Clin Invest. 1999;103:571–7. DOIPubMedGoogle Scholar
- Fong IW. Antibiotic effects in a rabbit model of Chlamydia pneumoniae-induced atherosclerosis. J Infect Dis. 2000;181(Suppl 3):514–8. DOIGoogle Scholar
- Laitinen K, Laurila A, Pyhälä L, Leinonen M, Saikku P. Chlamydia pneumoniae infection induces inflammatory changes in the aortas of rabbits. Infect Immun. 1997;65:4832–5.PubMedGoogle Scholar
- Muhlestein JB, Anderson JL, Hammond EH, Zhao L, Trehan S, Schwobe EP, Infection with Chlamydia pneumoniae accelerates the development of atherosclerosis and treatment with azithromycin prevents it in a rabbit model. Circulation. 1998;97:633–6.PubMedGoogle Scholar
- Blessing E, Lin T-M, Campbell LA, Rosenfeld M, Kuo C-C. Chlamydia pneumoniae induces inflammatory changes in hearts and aortas of C57BL/6J mice. Infect Immun. 2000;68:4765–8. DOIPubMedGoogle Scholar
- Moazed TC, Campbell LA, Rosenfeld ME, Grayston JT, Kuo CC. Chlamydia pneumoniae infection accelerates the progression of atherosclerosis in ApoE-deficient mice. J Infect Dis. 1999;180:238–41. DOIPubMedGoogle Scholar
- Hu H, Pierce GN, Zhong G. The atherogenic effects of chlamydia are dependent on serum cholesterol and specific to Chlamydia pneumoniae. J Clin Invest. 1999;103:747–53. DOIPubMedGoogle Scholar
- Blessing E, Campbell LA, Rosenfeld ME, Kuo C-C. Acceleration of the development of atherosclerosis following chronic infection with Chlamydia pneumoniae in cholesterol-fed C57BL/6J mice. Atherosclerosis. 2001. In press.PubMedGoogle Scholar
- Meier CR. Antibiotics in the prevention and treatment of coronary heart disease. J Infect Dis. 2000;181(Suppl 3):558–62. DOIPubMedGoogle Scholar
- Gupta S, Leatham EW, Carrington D, Mendall MA, Kaski JC, Camm AJ. Elevated Chlamydia pneumoniae antibodies, cardiovascular events and azithromycin in male survivors of myocardial infarction. Circulation. 1997;46:404–7.PubMedGoogle Scholar
- Gurfinkel E, Bozovich G, Beck E, Testa E, Livellara B, Mantner B. Treatment with the antibiotic roxithromycin in patients with acute non-Q-wave coronary syndromes. The final report of the ROXIS Study. Eur Heart J. 1999;20:121–7. DOIPubMedGoogle Scholar
- Muhlestein JB, Anderson JL, Carlquist JF, Salunkhe K, Horne BD, Pearson RR, Randomized secondary prevention trial of azithromycin in patients with coronary artery disease. Primary clinical results of the ACADEMIC study. Circulation. 2000;102:1755–60.PubMedGoogle Scholar
- Anderson JL, Muhlestein JB, Carlquist J. Randomized secondary prevention trial of azithromycin in patients with coronary artery disease and serological evidence for Chlamydia pneumoniae infection. The Azithromycin in Coronary Artery Disease: Elimination of Myocardial Infection with Chlamydia (ACADEMIC) study. Circulation. 1999;99:1540–7.PubMedGoogle Scholar
- Nieto FJ. Viruses and atherosclerosis: a critical review of the epidemiologic evidence. Am Heart J. 1999;138:S453–60. DOIPubMedGoogle Scholar
- Epstein SE, Zhou YF, Zhu JH. Infection and atherosclerosis: emerging mechanistic paradigms. Circulation. 1999;100:e20–8.PubMedGoogle Scholar
- Libby P, Egan D, Skarlatos S. Roles of infectious agents in atherosclerosis and restenosis: an assessment of the evidence and need for future research. Circulation. 1997;96:4095–103.PubMedGoogle Scholar
- Ridker PM, Hennekens CH, Stampfer MJ, Wang F. Prospective study of herpes simplex virus, cytomegalovirus, and the risk of future myocardial infarction and stroke. Circulation. 1998;98:2796–9.PubMedGoogle Scholar
- Sorlie PD, Nieto FJ, Adam E, Folsom AR, Shahar E, Massing M. A prospective study of cytomegalovirus, herpes simplex virus I, and coronary heart disease. Arch Intern Med. 2000;160:2027–32. DOIPubMedGoogle Scholar
- Adler SP, Hur JK, Wang JB, Vetrovec GW. Prior infection with cytomegalovirus is not a major risk factor for angiographically demonstrated coronary artery atherosclerosis. J Infect Dis. 1998;177:209–12. DOIPubMedGoogle Scholar
- Siscovick DS, Schwartz SM, Corey L, Grayston JT, Ashley R, Wang S-P, Chlamydia pneumoniae, herpes simplex virus type 1, and cytomegalovirus and incident myocardial infarction and coronary heart disease death in older adults: The Cardiovascular Health Study. Circulation. 2000;102:2335–40.PubMedGoogle Scholar
- Zhou YF, Leon MB, Waclawiw MA, Popma JJ, Yu ZX, Finkel T, Association between prior cytomegalovirus infection and the risk of restenosis after coronary atherectomy. N Engl J Med. 1996;335:624–30. DOIPubMedGoogle Scholar
- Smith K, Parsonnet J. Association between prior cytomegalovirus infection and the risk of restenosis after coronary atherosclerosis [Correspondence]. N Engl J Med. 1997;336:587. DOIPubMedGoogle Scholar
- Hosenpud JD. Coronary artery disease after heart transplantation and its relation to cytomegalovirus. Am Heart J. 1999;138(Suppl):S469–72. DOIPubMedGoogle Scholar
- Weis M, von Scheidt W. Cardiac allograft vasculopathy: a review. Circulation. 1997;96:2069–77.PubMedGoogle Scholar
- Koskinen O, Lemstrom K, Mattila S, Hayry P, Nieminen MS. Cytomegalovirus infection associated accelerated heart allograft arteriosclerosis may impair the late function of the graft. Clin Transplant. 1996;10:487–93.PubMedGoogle Scholar
- Chiu B. Multiple infections in carotid atherosclerotic plaques. Am Heart J. 1999;138:534–6. DOIPubMedGoogle Scholar
- Bartels C, Maass M, Bein G, Brill N, Bechtel JFM, Leyh R, Association of serology with the endovascular presence of Chlamydia pneumoniae and cytomegalovirus with coronary artery and vein graft disease. Circulation. 2000;101:137–41.PubMedGoogle Scholar
- Wu T-C, Hruban RH, Ambinder RF, Pizzorno M, Cameron DE, Baumgartner WA, Demonstration of cytomegalovirus nucleic acids in the coronary arteries of transplanted hearts. Am J Pathol. 1992;140:739–47.PubMedGoogle Scholar
- Gulizia JM, Kandolf R, Kendall TJ, Thieszen SL, Wilson JE, Radio SJ, Infrequency of cytomegalovirus genome in coronary arteriopathy of human heart allografts. Am J Pathol. 1995;147:461–75.PubMedGoogle Scholar
- Streblow DN, Soderberg-Naucler C, Vieira J, Smith P, Wakabayashi E, Ruchti F, The human cytomegalovirus chemokine receptor US28 mediates vascular smooth muscle cell migration. Cell. 1999;99:511–20. DOIPubMedGoogle Scholar
- Speir E, Modali R, Huang ES, Leon MB, Shawl F, Finfel T, Potential role of human cytomegalovirus and p53 interaction in coronary restenosis. Science. 1994;265:391–4. DOIPubMedGoogle Scholar
- Zhou YF, Shou M, Guetta E, Guzman R, Unger EF, Yu ZX, Cytomegalovirus infection of rats increases the neointimal response to vascular injury without consistent evidence of direct infection of the vascular wall. Circulation. 1999;100:1569–75.PubMedGoogle Scholar
- Persoons MC, Daemen MJ, van Kleef EM. Neointimal smooth muscle cell phenotype is important in its susceptibility to cytomegalovirus (CMV) infection: a study in rat. Cardiovasc Res. 1997;36:282–8. DOIPubMedGoogle Scholar
- Lemstrom K, Sihvola R, Bruggeman C, Hayry P, Koskinen P. Cytomegalovirus infection-enhanced cardiac allograft vasculopathy is abolished by DHPG prophylaxis in the rat. Circulation. 1997;95:2614–6.PubMedGoogle Scholar
- Genco RJ, Offenbacher S, Beck J, Rees T. Cardiovascular diseases and oral infections. In: Rose L, Genco R, Mealy BL, editors. Periodontal medicine. Hamilton, Ontario: B.C. Decker Corp; 2000. p. 63-82.
- Joshipura KJ, Douglass CW, Willett WC. Possible explanation for the tooth loss and cardiovascular disease relationship. Ann Periodontol. 1998;3:175–83. DOIPubMedGoogle Scholar
- Haraszthy VI, Zambon JJ, Trevisan M, Zeid M, Genco RJ. Identification of periodontal pathogens in atheromatous plaques. J Periodontol. 2000;71:1554–60. DOIPubMedGoogle Scholar
- Whincup P, Danesh J, Walker M, Lennon L, Thomson A, Appleby P, Prospective study of potentially virulent strains of Helicobacter pylori and coronary heart disease in middle-aged men. Circulation. 2000;101:1647–52.PubMedGoogle Scholar
- Nicholson AC, Hajjar DP. Herpesvirus in atherosclerosis and thrombosis: etiologic agents or ubiquitous bystanders? Arterioscler Thromb Vasc Biol. 1998;18:339–48. DOIPubMedGoogle Scholar
- Taylor C. Workshop on Emerging Issues in Microbial Infections and Cardiovascular Diseases, October 1998. NIAID Council News 1999;8:5. Available from: National Institute of Allergy and Infectious Diseases, National Institutes of Health: URL: http://www.niaid.nih.gov/ncn/newsletters/nl0199/page5.htm
- Zhu J, Nieto FJ, Horne BD, Anderson JL, Muhlestein JB, Epstein SE. Prospective study of pathogen burden and risk of myocardial infarction or death. Circulation. 2001;103:45–51.PubMedGoogle Scholar
- Wanishsawad C, Zhou YF, Epstein SE. Chlamydia pneumoniae-induced transactivation of the major immediate early promoter of cytomegalovirus: potential synergy of infectious agents in the pathogenesis of atherosclerosis. J Infect Dis. 2000;181:787–90. DOIPubMedGoogle Scholar
- Strong JP, Malcom GT, McMahan CA, Tracy RE, Newman WP III, Herderick EE, Prevalence and extent of atherosclerosis in adolescents and young adults: implications for prevention from the Pathobiological Determinants of Atherosclerosis in Youth Study. JAMA. 1999;281:727–35. DOIPubMedGoogle Scholar
- Williams KJ, Rodrigueza WV. Atherosclerosis: cell biology and proteins. Curr Opin Lipidol. 1998;9:511–3. DOIPubMedGoogle Scholar
- Barber DA, Burnett JC, Fitzpatrick LA, Sieck GC, Miller VM. Gender and relaxation to C-Type natriuretic peptide in porcine coronary arteries. J Cardiovasc Pharmacol. 1998;32:5–11. DOIPubMedGoogle Scholar
- Meydani M, Lipman RD, Han SN, Wu D, Beharka A, Martin KR, The effect of long-term supplementation with antioxidants. Ann N Y Acad Sci. 1998;854:352–60. DOIPubMedGoogle Scholar
- Hoogwerf BJ, Young JB. The HOPE study. Ramipril lowered cardiovascular risk, but vitamin E did not. Cleve Clin J Med. 2000;67:287–93.PubMedGoogle Scholar
- Stamm WE. Potential for antimicrobial resistance in Chlamydia pneumoniae. J Infect Dis. 2000;181(Suppl 3):456–9. DOIPubMedGoogle Scholar
- Seppälä H, Klaukka T, Vuopio-Varkila J, Muotiala A, Helenius H, Lager H, The effect of changes in the consumption of macrolide antibiotics on erythromycin resistance in group A streptococci in Finland. Finnish Study Group for antimicrobial resistance. N Engl J Med. 1997;337:441–6. DOIPubMedGoogle Scholar
- Arason VA, Kristinsson KG, Sigurdsson JA, Stefansdottir G, Molstad S, Gudmundsson S. Do antimicrobials increase the carriage rate of penicillin resistant pneumococci in children? Cross-sectional prevalence study. BMJ. 1996;313:387–91. DOIPubMedGoogle Scholar
- Leach AJ, Shelby-James TM, Mayo M, Gratten M, Laming AC, Currie BJ, A prospective study of the impact of community-based azithromycin therapy on trachoma carriage and resistance of S. pneumoniae. Clin Infect Dis. 1997;24:356–62. DOIPubMedGoogle Scholar
- Dagan R, Leibovitz E, Greenberg D, Yagupsky P, Fliss DM, Leiberman A. Dynamics of pneumococcal nasopharyngeal colonization during the first days of antibiotic treatment in pediatric patients. Pediatr Infect Dis J. 1998;17:880–5. DOIPubMedGoogle Scholar
- Stephens RS. Chlamydial genomics and vaccine antigen discovery. J Infect Dis. 2000;181(Suppl 3):521–3. DOIPubMedGoogle Scholar
- Murdin AD, Dunn P, Sodoyer R, Wang J, Caterini J, Brunham RC, Use of mouse lung challenge model to identify antigens protective against Chlamydia pneumoniae lung infection. J Infect Dis. 2000;181(Suppl 3):544–51. DOIPubMedGoogle Scholar
- Craighead JE. Report of a workshop: disease accentuation after immunization with inactivated microbial vaccines. J Infect Dis. 1975;131:749–54. DOIPubMedGoogle Scholar
- Bachmaeir K, Neu N, de la Maza LM, Pal S, Hessel A, Penninger JM. Chlamydia infections and heart disease links through antigenic mimicry. Science. 1999;283:1335–9. DOIPubMedGoogle Scholar
- Dodet B, Plotkin SA, Ross R, Falk E, Kritchevsky D, Valtonen VV, Proceedings of an International Symposium on Infection and Atherosclerosis. Les Pensieres, Veyrier-du-Lac, France, December 6-9, 1998. Am Heart J. 1999;138:S417–560. DOIPubMedGoogle Scholar
- Gilbert DN, Ewald PW, Cochran GM, Grayston JT, Saikku P, Leinonen M, Preface. J Infect Dis. 2000;181(Suppl 3):S393–586. DOIPubMedGoogle Scholar
Figures
Cite This ArticleTable of Contents – Volume 7, Number 5—October 2001
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Siobhán O'Connor; Centers for Disease Control and Prevention, 1600 Clifton Rd. NE, Mailstop C-12, Atlanta, GA 30333, USA; fax: 404-639-3039
Top