Volume 9, Number 7—July 2003
Research
Molecular Epidemiology of O139 Vibrio cholerae: Mutation, Lateral Gene Transfer, and Founder Flush
Cite This Article
Citation for Media
Abstract
Vibrio cholerae in O-group 139 was first isolated in 1992 and by 1993 had been found throughout the Indian subcontinent. This epidemic expansion probably resulted from a single source after a lateral gene transfer (LGT) event that changed the serotype of an epidemic V. cholerae O1 El Tor strain to O139. However, some studies found substantial genetic diversity, perhaps caused by multiple origins. To further explore the relatedness of O139 strains, we analyzed nine sequenced loci from 96 isolates from patients at the Infectious Diseases Hospital, Calcutta, from 1992 to 2000. We found 64 novel alleles distributed among 51 sequence types. LGT events produced three times the number of nucleotide changes compared to mutation. In contrast to the traditional concept of epidemic spread of a homogeneous clone, the establishment of variant alleles generated by LGT during the rapid expansion of a clonal bacterial population may be a paradigm in infections and epidemics.
An epidemic of cholera began in Madras, India, in 1992 and within a year had spread across the Indian subcontinent, with cases numbering in the millions (1,2). Vibrio cholerae isolates from this epidemic had a previously unidentified serotype, subsequently designated as O139 Bengal (1,2). This new serotype appears to have resulted when a lateral gene transfer (LGT) event occurred that replaced the 22 kb of the wbf region (encoding the O1 antigen) of a seventh pandemic V. cholerae O1 El Tor strain with a 37-kb region encoding the O139 polysaccharide (3–5). The epidemic spread rapidly through all age groups, as persons with previous exposure to V. cholerae O1 were not immune to O139 infection. Since 1992, O139 strains have established endemicity in this geographic region and account for a variable percentage of cholera cases every year (6).
Genetic variation observed in O139 isolates has been attributed to many causes. Variation in restriction fragment length polymorphism (RFLP) analysis of rDNA genes (7) and in recA sequence (8) has been interpreted as evidence for multiple origins. Genetic variability in RFLP of the CTX element (6) has been attributed to phage-mediated recombination. Variation in antimicrobial susceptibility (9) has been attributed to plasmid exchange in response to selective pressure from drug use. The variation in pulsed-field gel electrophoresis (PFGE) analysis of genomic restriction fragments (6,10) has been attributed to point mutations. Multilocus sequence typing (MLST), which has been used in the evaluation of a number of other bacterial species (11–14), provides an alternative method for measuring genetic relatedness and has provided data for identifying both point mutations and LGT events (14). MLST has improved discriminatory power over PFGE in some cases, e.g., Enterococcus (15) and Salmonella (16); however, in the case of Escherichia coli O157, it does not because of an absence of sequence variation in the clonally derived isolates (17). A small MLST study of O139 isolates of V. cholerae did not identify any LGT events (18).
To understand the evolutionary dynamics of V. cholerae O139, we sequenced segments from nine loci, including seven that may be classified as traditional housekeeping genes, one that carries the genes for cholera toxin, and another that is next to the insertion sequence within the O139 wbf region (3–5). Thus, the last two loci might be expected to show LGT, because they are associated with known mobile elements, but the other seven loci would not be expected to show LGT. However, we found putative LGT alleles at all nine loci in the 96 clonally related O139 isolates.
We evaluated nine loci—dnaE, lap, recA, pgm, gyrB, cat, chi, rstR, and gmd—from 96 V. cholerae O139 isolated from patients seen at the Infectious Diseases Hospital, Calcutta, from 1992 to 2000 (see Appendix, online only). DNA was prepared from overnight cultures by using PrepMan Ultra (Applied Biosystems Inc., Foster City, CA) at the University of Maryland School of Medicine. Each locus was amplified by using polymerase chain reaction (PCR) with primers (Table 1) selected from a conserved region of the locus, as determined by aligning sequences from GenBank. Our primers selectively amplified the original O139 rstR gene found in all isolates and not the additional one found in some recently inserted CTX elements (19). The presence of amplified products was confirmed on agarose gels. Purification of the products was performed by using Millipore filters. The purified PCR products were sequenced in both directions by using the same primers used for amplification and Big Dye cycle sequencing kit (ABI) in accordance with manufacturer’s instructions. The fluorescently labeled products were separated and detected by using either an ABI 377 or 3700 Automatic Sequencer (ABI). The trace files were read by using Phred (20,21) and Phrap (22). Low-quality sequence at the ends was trimmed, and the contigs from each individual isolate were aligned by using Clustal X (23). Variable nucleotides were identified manually. Isolates with identical alleles were identified from a distance matrix obtained from PAUP (24). The alleles have been assigned GenBank accession numbers AY297845 to AY297921.
Figure 1

Figure 1. Bar graph of the number of novel alleles (y-axis) with a specific number of nucleotide differences from the ancestral allele. Two alleles with 24-bp and 113-bp differences are excluded from the...
The expected number of alleles that were a result of point mutations was calculated. All point mutations were assumed to occur independently; thus, the expected number of alleles with >2 nucleotides (nt) can be calculated, and the excess number of observed alleles was attributed to conspecific LGT of homologous genes. If one assumes that p is the probability of seeing a single mutation in an allele, the chance of seeing two mutations on the same allele is p2; the probability of seeing three or more mutations is p3. Probability can be calculated from the data by dividing the number of alleles with a single nucleotide difference, 34, by 785, the number of alleles in which observing a point mutation is possible (the 6-bp deletion; the recombinant gmd, recA alleles; and all duplicate novel alleles were excluded). Thus, p equals 0.043, p2 equals 0.0018, and p3 equals 8 x 10-5. When these probabilities are multiplied by the total number of alleles, 785, the expected number of alleles containing two independent point mutations is 1.45, and the expected number containing three or more is 0.06 (Figure 1).
Each of the loci examined had a variable number of observed alleles: 9 for dnaE, 20 for lap, 11 for rstR, 11 for gmd, 2 for recA, 8 for pgm, 4 for gyrB, 7 for cat, and 5 for chi. The most variable, lap with 20 alleles, was expected because it is a highly variable locus when analyzed with multilocus enzyme electrophoresis (25). The most common allele was present in 91% of isolates (n=87) for dnaE, 77% (n=86) for lap, 79% (n=90) for rstR, 82% (n=87) for gmd, 99% (n=96) for recA, 90% (n=94) for pgm, 97% (n=92) for gyrB, 93% (n=88) for cat, and 94% (n=89) for chi. Thus, the pattern for each locus consists of a common or ancestral allele and a series of rare alleles, as expected for the expansion of a clone.
Of the 64 less frequent alleles, some result from LGT and others from mutation. The three alleles with the largest changes are unlikely to be due to point mutations. First, a gmd allele that differed by 113 of the 360 bp sequenced, when compared with sequences in GenBank using BLAST (available from: URL: www.ncbi.nlm.nih.gov/BLAST/) showed greater similarity to gmd from E. coli (AF061251) than gmd from V. cholerae, consistent with LGT of a homologous gene into the V. cholerae genome. Second, an alternative recA allele that differs by 24 nt is likely to be the result of LGT of a homologous gene. Although substantial, the number of nucleotide differences is not large enough for the allele to be clustered with sequences from V. mimicus, the closest sibling species to V. cholerae (8), a finding that suggests that recombination occurred within V. cholerae. Third, a lap allele had a 6-bp deletion and a single nucleotide difference that may be the result of a double-strand break repair.
We calculated that at least 26 putative conspecific LGT events occurred in the 96 isolates studied. Figure 1 shows the number of nucleotide differences between each novel allele and the ancestral allele. If all point mutations are assumed to occur independently, the expected number of alleles with two or more variable nucleotides can be calculated and the excess number of observed alleles attributed to conspecific LGT of homologous genes. If all point mutations are assumed to occur independently, the expected number of alleles with two or more variable nucleotides can be calculated and the excess number of observed alleles attributed to conspecific recombination of homologous genes. The expected number of alleles containing two independent point mutations is 1.45, and the expected number containing three or more is 0.06. Since 11 alleles were observed with 2 nt differences, 9 more than expected, and 16 were observed with >3 differences, 16 more than expected, all of these alleles probably did not occur through mutation; more likely, these alleles are the result of LGT. Thus, we would estimate that 26 alleles (9 + 16 + recA allele above) are putatively due to conspecific LGT of homologous genes.
The putative conspecific LGT alleles, although fewer in number (26 alleles) than the assumed number of mutation-derived alleles (34 alleles), provide most of the nucleotide differences between alleles. The 120 nt changes introduced by conspecific LGT events are approximately three times the 38 (34 single mutations + 4 2x2 double mutations) introduced by mutation. This calculation is conservative: The 26 conspecific LGT events may represent an underestimate of the number because some of the alleles differing by <1 nt may have resulted from LGT.
The analysis of all nine loci from each isolate was based on the sequence type (ST). Each isolate was defined by a 9-digit number composed of the assigned allele number at each of the nine loci in the following order: dnaE, lap, rstR, gmd, recA, pgm, gyrB, cat, and chi. The most common allele was arbitrarily assigned as number 1. Thus, the ST of all the most common alleles is ST 1,1,1,1,1,1,1,1,1. Missing data were assigned the most common allele. This assumption is conservative, minimizes the observed amount of variation, and is consistent with the preponderance of common alleles found at each locus.
Fifty-one unique STs were found in the 96 isolates tested, reflecting relatively extensive genetic diversity. The overall average of 0.53 unique STs per isolate examined is similar to that seen in every year including 1992 (Table 2). Six STs occur more than once. As expected, the ancestral ST:1,1,1,1,1,1,1,1,1, found in 40 isolates, occurred in all years. Among the others, ST:1,1,2,1,1,1,1,1,1 was found three times, once each in 1995, 1996, and 1997. ST:1,2,1,1,1,1,1,1,1 and ST:1,1,7,1,1,1,1,1,1 were found twice in 1992 and 1994, respectively. ST 1,1,1,6,1,1,1,1,1 was found once in 1998 and again in 1999. ST:1,1,1,1,1,4,1,1,1 was found in 1995 and 1998. Since the number of STs is large (51 types), and number of samples in a collection period is small (8–13 samples; Table 2), STs seen in multiple years must not only persist but also represent a substantial portion of the epidemic O139 V. cholerae population.
Figure 2
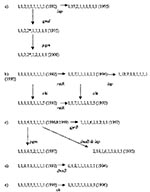
Figure 2. Five groups of related sequence types of Vibrio cholerae O139.
Five of the novel STs are related to other novel STs by allelic change at another second or third locus. One sequence type evolved into three related types found in subsequent years (Figure 2a). The starred gmd allele is one related to the E. coli sequence, and its presence in two distinct related STs in two different years demonstrates its establishment in the population. That the pattern seen in Figure 2b of ancestral alleles rstR 1 and chi 1 and two variant alleles, rstR 7 and chi 5, was found in all combinations is indicative of an LGT event. Figure 2c-e shows three additional groups of related sequences. In Figure 2a, b, and d, the ST with the larger number of novel alleles occurred in later years. In contrast, in Figure 2c and e, the ST with the larger number of novel alleles occurred in the earlier years. The lack of an overall temporal relationship may result from the small sample size (8–13 isolates) in any year.
One isolate, CRC5, is unusual because it has no sequenced alleles of the ancestral type. Nevertheless, the alleles from this isolate are closely related to those of the ancestral type. Each CRC5 allele differs from the ancestral allele by 7 nt for dnaE, 3 nt for lap, 4 nt for rstR, 24 nt for recA, 6 nt for pgm, 10 nt for gyrB, and 4nt for chi. A comprehensive survey of the genetic distances for these loci could determine the average distance between alleles for each of these loci. The data would provide insight into whether this isolate represents a second derivation of the O139 clinical type from an environmental strain or if it is a genetic outlier within the clonally related, but diversified, O139 epidemic type.
The emergence and pandemic spread of V. cholerae O139 Bengal represented a chance to examine evolution of a bacterial strain in the midst of a clonal expansion. Our results are consistent with clonal expansion and subsequent divergence as described by Spratt and Maiden (26). Putative recombinant alleles were found at all nine loci among the 96 clonally related O139 isolates. One gmd allele from V. cholerae was most similar to a gmd allele from E. coli. The number of base-pair differences among other alleles was higher than expected on the basis of a simple computation for the accumulation of independent mutations. This finding suggests that many of these events were due to LGT. When we applied our criteria to the novel alleles identified in a previous study (18), 11 of the 13 would be considered to have resulted from LGT, since the number of nucleotide differences to the ancestral allele varied from 4 to 19. Thus, for V. cholerae, like Neisseria, Streptococcus, and other bacterial species (11–14), conspecific recombination of homologous genes appears to be common and responsible for most of the alleles with multiple nucleotide differences and the majority of the nucleotide differences. The genetic variability at the nine loci alters our understanding of evolution in bacteria, showing that recombination in V. cholerae occurs frequently and most nucleotide changes occur by means of a recombination that can alter any gene.
The proportion of recombinants from conspecific recombination, 3.5% (28/785) is greater than that from transgeneric recombination (0.01% from the acquisition of E. coli gmd by one isolate). One potential implication of a greater rate of conspecific recombination may be that, over time, it will maintain the species identity of each individual bacterium, despite the constant bombardment of homologous genes from other genera. Although at first glance the frequency of the novel sequence types appears to conflict between our study and an earlier study (18), the observations may be reconciled on the basis of both the observed frequencies and the timing of the observations. Both studies identified a common ancestral allele in from 77% to 99% of isolates in our study and a series of rare alleles with 1–19 variant alleles for each locus. These studies reported 10% novel sequence types in 29 isolates that were collected from “the first epidemic period,” from 1992 to 1993 (18). Our data from 1992 showed 33% novel sequence types from a sample of nine. These data are not statistically different (chi-square test=2.4, p=0.12). However, the researchers’ estimate of frequency (18) is more likely to be correct because of the larger sample size. The dates of collection may also be important because our collection of isolates from 1993 began in March, when the number of O139 cases at the Infectious Diseases Hospital rose from <10 to >80 per month, corresponding to a rapid population expansion or flush. Thus, we can predict that we would see substantial variation in our sample.
The genetic diversity was greater in the V. cholerae O139 isolates than in other clinically associated clones. In V. parahaemolyticus O3:K6, a pandemic strain, 94% of strains were identical at four loci (N. Chowdhury et al., unpub. data). In E. coli O157, all 77 isolates were identical at seven loci in spite of variation between isolates on PFGE (17). Although V. parahaemolyticus and E. coli are widespread pathogens, they differ from V. cholerae O139 because their population size has expanded much more slowly.
Among O139 isolates, the substantial genetic diversity found in the first year of the epidemic may reflect a “founder flush” phenomenon. During times of population expansion, i.e., a flush, any novel genotype with similar or even slightly deleterious fitness compared to the founder genotype will produce sufficient offspring to become established in the population (27). A founder flush appears to have occurred in the establishment of Helicobacter pylori in a single person (28). Although other previous descriptions of this phenomenon have been limited to insects, specifically butterflies (29) and drosophilids (30), we believe that the founder flush phenomenon may become the paradigm for epidemic bacterial expansion in individual patients and populations. This founder flush phenomenon, in turn, has implications for our interpretation of “clonality” among epidemic isolates and for our understanding of factors that contribute to the emergence of new pathogenic strains.
Dr. Garg completed this work as part of her Ph.D. dissertation at University of Calcutta. She is a postdoctoral fellow at the University of Maryland School of Medicine, pursuing studies of thrombospondin in human microvascular endothelial cells.
Acknowledgment
This work was supported in part by an American Society for Microbiology travel fellowship to Dr. Garg.
References
- Bhattacharya SK, Bhattacharya MK, Nair GB, Dutta D, Deb A, Ramamurthy T, Clinical profile of acute diarrhoea cases infected with the new epidemic strain of Vibrio cholerae O139: designation of the disease as cholera. J Infect. 1993;27:11–8. DOIPubMedGoogle Scholar
- Nair GB, Ramamurthy SK, Mukhopadhyay T, Garg S, Bhattacharya MK, Takeda T, Spread of Vibrio cholerae O139 Bengal in India. J Infect Dis. 1994;169:1029–34.PubMedGoogle Scholar
- Comstock LE, Johnson JA, Michalski JM, Morris JG, Kaper JB. Cloning and sequence of a region encoding surface polysaccharide of Vibrio cholerae O139 and characterization of the insertion site in the chromosome of Vibrio cholerae O1. Mol Microbiol. 1966;19:815–26. DOIPubMedGoogle Scholar
- Stroeher UH, Parasivam G, Dredge BK, Manning PA. Novel Vibrio cholerae O139 genes involved in lipopolysaccharide biosynthesis. J Bacteriol. 1997;179:2740–7.PubMedGoogle Scholar
- Bik EM, Bunschoten AE, Willems RJL, Chang ACY, Mooi FR. Genetic organization and functional analysis of the otn DNA essential for cell-wall polysaccharide synthesis in Vibrio cholerae O139. Mol Microbiol. 1996;20:799–811. DOIPubMedGoogle Scholar
- Basu A, Garg P, Datta S, Chakraborty S, Bhattacharya T, Yamasaki S, Vibrio cholerae O139 in Calcutta, 1992–1998: incidence, antibiograms, and genotypes. Emerg Infect Dis. 2000;6:139–47. DOIPubMedGoogle Scholar
- Faruque SM, Saha MN, Asadulghani, Bag PK, Bhadra RK, Bhattacharya SK, et al. Genomic diversity among Vibrio cholerae O139 strains isolated in Bangladesh and India between 1992 and 1998. FEMS Microbiol Lett. 2000;184:279–84. DOIPubMedGoogle Scholar
- Stine OC, Sozhamannan S, Gou Q, Zheng S, Morris JG, Johnson JA. Phylogeny of Vibrio cholerae based on recA sequence. Infect Immun. 2000;68:7180–5. DOIPubMedGoogle Scholar
- Garg P, Chakraborty S, Basu I, Datta S, Rajendran K, Bhattacharya T, Expanding multiple antibiotic resistance among clinical strains of Vibrio cholerae isolated from 1992–7 in Calcutta, India. Epidemiol Infect. 2000;124:393–9. DOIPubMedGoogle Scholar
- Kurazono H, Yamasaki S, Ratchtrachenchai O, Nair GB, Takeda Y. Analysis of Vibrio cholerae O139 Bengal isolated from different geographical areas using macrorestriction DNA analysis. Microbiol Immunol. 1996;40:303–5.PubMedGoogle Scholar
- Maiden MCJ, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A. 1998;95:3140–5. DOIPubMedGoogle Scholar
- Dingle KE, Colles FM, Wareing DR, Ure R, Fox AJ, Bolton FE, Multilocus sequence typing system for Campylobacter jejuni. J Clin Microbiol. 2001;39:14–23. DOIPubMedGoogle Scholar
- Enright MC, Day NP, Davies CE, Peacock SJ, Spratt BG. Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J Clin Microbiol. 2000;38:1008–15.PubMedGoogle Scholar
- Feil EJ, Smith JM, Enright MC, Spratt BG. Estimating recombinational parameters in Streptococcus pneumoniae from multilocus sequence typing data. Genetics. 2000;154:1439–50.PubMedGoogle Scholar
- Nallapareddy SR, Duh RW, Singh KV, Murray BE. Molecular typing of selected Enterococcus faecalis isolates: pilot study using multilocus sequence typing and pulsed-field gel electrophoresis. J Clin Microbiol. 2002;40:868–76. DOIPubMedGoogle Scholar
- Kotetishvili M, Stine OC, Kreger A, Morris JG Jr, Sulakvelidze A. Multilocus sequence typing for characterization of clinical and environmental Salmonella strains. J Clin Microbiol. 2002;40:1626–35. DOIPubMedGoogle Scholar
- Noller AC, McEllistrem MC, Stine OC, Morris JG, Boxrud DJ, Dixon B, Lack of DNA sequence diversity among Escherichia coli O157:H7 isolates that are distinct by pulsed-field gel electrophoresis. J Clin Microbiol. 2003;41:675–9. DOIPubMedGoogle Scholar
- Farfan M, Minana-Galbis D, Fuste MC, Loren JG. Allelic diversity and population structure in Vibrio cholerae O139 Bengal based on nucleotide sequence diversity. J Bacteriol. 2002;184:1304–13. DOIPubMedGoogle Scholar
- Mukhopadhyay AK, Chakraborty S, Takeda Y, Nair GB, Berg DE. Characterization of VPI pathogenicity island and CTXΦ prophage in environmental strains of Vibrio cholerae. J Bacteriol. 2001;183:4737–46. DOIPubMedGoogle Scholar
- Ewing B, Greene P. Base-calling of automated sequencer traces using Phred. II. Error probabilities. Genome Res. 1998;8:186–94.PubMedGoogle Scholar
- Ewing B, Hillier L, Wendl M, Greene P. Base-calling of automated sequencer traces using Phred. I. Accuracy assessment. Genome Res. 1998;8:175–85.PubMedGoogle Scholar
- Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ. Multiple sequence alignment with Clustal X. Trends Biochem Sci. 1998;23:403–5. DOIPubMedGoogle Scholar
- Swofford D. PAUP* beta version. Sunderland (MA): Sinauer Assoc; 2000.
- Beltran P, Delgrado G, Navarro A, Trujillo F, Selander RK, Cravioto A. Genetic diversity and population structure of Vibrio cholerae. J Clin Microbiol. 1999;37:581–90.PubMedGoogle Scholar
- Spratt BG, Maiden MCJ. Bacterial population genetics, evolution and epidemiology. Philos Trans R Soc Lond Biol Sci. 1999;354:701–10. DOIPubMedGoogle Scholar
- Wallace B. Basic populations genetics. New York: Columbia University Press; 1981. p. 429–32.
- Israel DA, Salama N, Krishna U, Rieger UM, Atherton JC, Falkow S, Helicobacter pylori genetic diversity within the gastric niche of a single host. Proc Natl Acad Sci U S A. 2001;98:14625–30. DOIPubMedGoogle Scholar
- Ford HD, Ford EB. Fluctuation in numbers, and its influence on variation, in Melitaea aurina, Rott. (Lepidoptera). Transactions of the Royal Entomological Society London 1930;78:345–51.
- Carson HL. The population flush and its genetic consequences. In: Lewontin RC, editor Population biology and evolution. Syracuse (NY): Syracuse University Press;1968. p. 123–37.
Figures
Tables
Cite This ArticleTable of Contents – Volume 9, Number 7—July 2003
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
O. Colin Stine, Department of Epidemiology and Preventive Medicine, University of Maryland School of Medicine, Baltimore, MD 21201, USA; fax: 410-706-1644
Top