Volume 32, Number 4—April 2026
Research
Geographically Distinct Circulation of Genotype II and III St. Louis Encephalitis Virus, Texas, USA, 2009–2024
Alexander R. Kneubehl, Daniel P. Rehm, Michael W. Curtis, Bianca M. Wimmer, Bethany Bolling, Angie Broussard, Jeremy Vela, Jennifer Rocha, Lindsey Templeton, Maximea Vigilant, Courtney Standlee, Steven M. Presley, Job E. Lopez, and Shannon E. Ronca
Figure 3
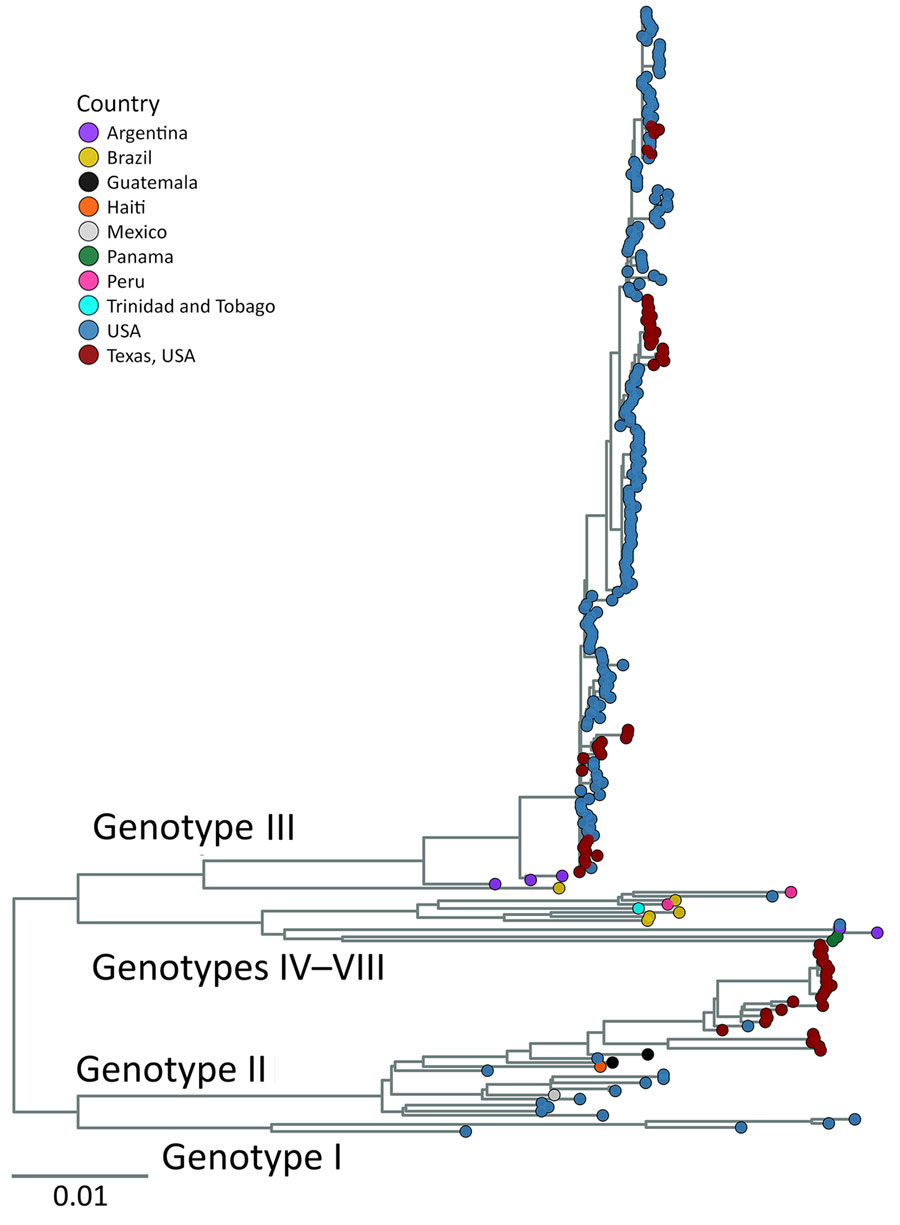
Figure 3. Maximum-likelihood phylogenic analysis of all St. Louis encephalitis virus genomes from study of circulation of genotype II and III St. Louis encephalitis virus, Texas, USA, 2009–2024. Maximum-likelihood inferred tree shows 276 genomes currently available that have <30% of the genome missing. Tip color represents sample’s country of origin; samples from Texas are labeled with a separate color (red) to distinguish Texas samples from rest of United States. Genotypes annotated on the tree indicate which clade contains which genotype or genotypes. Tree was midpoint rooted. Scale bar indicates number of nucleotide substitutions per site.
Page created: March 04, 2026
Page updated: April 15, 2026
Page reviewed: April 15, 2026
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.