Volume 12, Number 9—September 2006
Research
Differentiation of Tuberculosis Strains in a Population with Mainly Beijing-family Strains
Cite This Article
Citation for Media
Abstract
A high prevalence of tuberculosis (TB) isolates that are genetically homogenous and from the Beijing family has been reported in Russia. To map TB transmission caused by these strains, new genotyping systems are needed. Mycobacterial interspersed repetitive units (MIRUs) offer the possibility of rapid PCR-based typing with comparable discrimination to IS6110 restriction fragment length polymorphism techniques. Spoligotyping and detection of IS6110 insertion in the dnaA-dnaN region were used to identify Beijing strains in 187 Mycobacterium tuberculosis isolates from Samara, Russia. The Beijing isolates were analyzed by using 12-MIRU and 3–exact tandem repeats (ETR) loci and by an expanded set of 10 additional variable number tandem repeats loci. The expanded set of 25 MIRUs provided better discrimination than the original set of 15 (Hunter-Gaston diversity index 0.870 vs 0.625). Loci MIRU 26, 1982, and 3232 were the most polymorphic in Beijing isolates.
Rising rates of tuberculosis (TB) (1) and increasing resistance of drugs are substantial barriers to successful patient management and TB control programs. Russia, named by the World Health Organization as one of the 22 countries with the highest TB prevalence, has seen increasing rates of TB and HIV during the past decade (2–6) and high levels of resistance, particularly to multiple drugs (7,8).
The high discriminatory power of restriction fragment length polymorphism (RFLP) analysis based on the insertion sequence IS6110 has provided the backbone of these analyses (9). Unfortunately, the technique is time-consuming, technically demanding, and insufficiently discriminating when used alone with isolates containing <5 IS6110 sequences in the genome (10,11). Isolates with low numbers of copies account for as much as 20% of TB isolates in some populations (12).
Techniques based on PCR amplification of repetitive sequences are more rapid than RFLP, but their discriminatory power is usually lower. Among these techniques, spoligotyping is widely used to differentiate strains belonging to the Mycobacterium tuberculosis complex. Spoligotyping has been particularly useful for identifying strains belonging to the Beijing/W family of M. tuberculosis because of the characteristic spoligotyping pattern with the absence of spacers 1–34 in the direct repeat (DR) region of the M. tuberculosis genome (13). Beijing family strains are dominant across many Asian and former Soviet Union countries, and W strains are responsible for outbreaks of multidrug-resistant TB in the United States. These strains are now considered to be members of the same phylogenetic lineage, sharing key characteristics such as a similar RFLP pattern of 15–26 bands, IS6110 insertions in the dnaA-dnaN and NTF-1 chromosomal regions, a characteristic pattern of single nucleotide polymorphism, and a spoligotyping pattern with the presence of spacers 35–43 and absence of spacers 1–34 in the DR region of the M. tuberculosis genome (14–17). Detecting the IS6110 insertion in the dnaA-dnaN intergenic region may also identify the Beijing/W genotype (14,15).
Several studies have shown that a high proportion of TB isolates in Russia (particularly those that are drug resistant) belong to the Beijing family (7,8,18). To assess TB transmission in Russia, any genotyping system must be able to discriminate among Beijing strains. The identification of variable number tandem repeats (VNTRs) (19) in M. tuberculosis has offered the possibility of rapid amplification-based techniques with comparable discrimination to RFLP-IS6110 typing (20–23).
As with other PCR-based genotyping techniques, VNTR analysis uses small quantities of crude bacterial lysates; it is less labor-intensive than RFLP-IS6110 typing, and automation is relatively straightforward (22). Moreover, determination of a limited number of polymorphic loci can provide sufficient discriminative power for a given local population and may increase the cost-effectiveness of molecular typing. Several panels of M. tuberculosis VNTRs have been used with some success previously: exact tandem repeats (ETRs) (19), MIRUs (21,22), and 2 panels of loci known as QUB and Mtub (24–27). Our aim was to determine the discriminative power of an expanded set of 25 VNTR loci when applied to TB strains in Russia where Beijing strains dominate.
M. tuberculosis Strains
A total of 187 M. tuberculosis strains were analyzed. They were selected from 880 M. tuberculosis strains isolated from patients (1 isolate per patient) with radiologically confirmed pulmonary TB, identified from all TB treatment facilities across Samara Oblast in central Russia (12 civilian TB hospitals and dispensaries and 1 prison TB hospital) during 2001–2002. The test panel included 138 (33.7%) of 409 strains isolated from patients in the civilian TB hospitals and dispensaries and 49 (10.4%) of 471 isolates from prisoners. Within sets of cultures isolated from civilians, every third isolate was selected for this study. Because isolates from prisoners were overrepresented, every 10th isolate was selected. The representation of isolates in the test panel was approximately proportional to the number of TB isolates from each clinical site. In the Samara region, the incidence of TB at the time of the study was 86.1/100,000 population.
Patient populations are described in previous publications (18,28). All patients with TB are tested for HIV as a part of routine practice in Russia. History of vaccination with M. bovis BCG was confirmed by presence of an appropriate scar.
Molecular Epidemiologic Analysis
Crude DNA extracts were obtained by heating cell suspensions with chloroform at 80°C as previously described (29). Spoligotyping used a standardized method (30) to identify isolates belonging to the Beijing family; results were confirmed by analysis of the dnaA to dnaN region in all isolates. The IS6110 insertion in the origin of replication was detected between dnaA and dnaN genes as described previously (15,16). Briefly, PCR was performed in a 20-μL volume of 2 μL 10× PCR buffer (Bioline Ltd, London, UK); 0.5 units Taq polymerase (Bioline Ltd); 0.5 μL 2 mmol dNTP mixture (Bioline Ltd); 0.5 μL 20-μmol mix of forward and reverse primers, 15.5 μL water, and 1 μL of crude DNA extract. Thermal cycling was performed on a PerkinElmer 9700 thermocycler (PerkinElmer, Warrington, UK) as follows: 4 min at 94°C; 30 cycles of 30 s at 94°C, 30 s at 60°C, and 2 min at 72°C; followed by 7 min at 72°C and holding at 4°C. Amplification products were analyzed by electrophoresis on 1.5% agarose gel. Strains with insertion (i.e., Beijing family) yielded a product of ≈2 kb, and fragments of ≈550 bp (no insertion) indicated strains other than Beijing.
All 187 TB isolates were tested by using the set of 12-MIRU loci and the 3-ETR (A, B, and C) loci (19,22) (nos. 1–15, Table A1). Beijing isolates were further analyzed by using an additional panel of VNTR loci (0424, 0531, 1955, 1982, 2074, 2163a, 3232, 3239, 3336, and 3690, Table A1). Primers for loci at which predicted fragment size would exceed 1 kb were redesigned to enable analysis with the CEQ8000 equipment (Beckman Coulter, Fullerton, CA, USA).
Multiplex and simplex PCRs were performed, taking into consideration dye labeling and expected length of PCR products. Simplex PCR was used to amplify fragments in loci MIRU 20, ETR-C, MIRU 26, VNTR 424, VNTR 531, VNTR 1955, VNTR 1982, VNTR 2163a, VNTR 3232, VNTR 3239, and VNTR 3336. For other loci, multiplex PCR mixtures were prepared as follows: set 1 contained MIRU 4 and MIRU 16, set 2 contained MIRU 39 and ETR-A, set 3 contained MIRU 2 and MIRU 24, set 4 contained MIRU 31 and MIRU 40, set 5 contained MIRU 10 and MIRU 23, set 6 contained MIRU 27 and ETR-B, and set 7 contained VNTR 2074 and VNTR 3690.
For all mixtures, PCR was performed in 10-μL volumes containing 1 μL 10× PCR buffer (containing 1.5 mmol/L MgCl2, Bioline Ltd); 0.5 U Taq polymerase (Bioline Ltd); 0.25 μl 2-mmol dNTP mixture (Bioline Ltd); 0.5 μL 20-μmol mixture of forward and reverse primers as described above, 7.0 μL water, and 1 μL DNA extract. For loci VNTR 424, VNTR 531, VNTR 1955, VNTR 1982, VNTR 2074, VNTR 2163a, VNTR 3232, VNTR 3239, VNTR 3336, and VNTR 3690, the mixture also contained 0.5 μL dimethylsulfoxide (Sigma, Dorset, UK). Thermal cycling programs were identical for all loci, and thermal cycling was performed on a PerkinElmer 9700 thermocycler by using the following parameters: 3 min at 95°C; 30 cycles of 30 s at 95°C, 30 s at 60°C, and 60 s at 72°C; followed by 5 min at 72°C.
Automated analysis of PCR fragments length was performed by using a Beckman Coulter CEQ8000 automatic sequencer. Multiple PCR products were analyzed in capillaries as follows: capillary 1 contained PCR products for loci 2, 4, 10, 16, 23, and 24; capillary 2 for loci 27, 31, 39, ETR-A, and ETR-B; capillary 3 for loci 20, 26, and ETR-C; capillary 4 for loci 0424, 0531, and 1955; capillary 5 for loci 2974, 3232, 3239, and 3690; and capillary 6 for loci 1982, 2163a, and 3336.
Before being loaded onto the sequencer, PCR products were diluted in purified water (Sigma) as follows: products labeled with dye 2 were diluted to 10-fold; with dye 3, to 30-fold; and with dye 4, to 60-fold. A total of 1 μL of diluted PCR products mixture was added to 25 μL formamide (Beckman Coulter) containing 0.1 μL of DNA size standard 600 (Beckman Coulter), and 0.1 μL DNA size standard 640–1,000 (Bio Ventures Inc., Murfreesboro, TN, USA), labeled with dye 1. Fragment length estimation was performed by using proprietary software (Beckman Coulter).
Figure
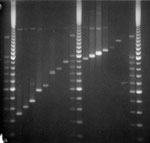
Figure. PCR analysis of VNTR3232 locus. Lane 1, 2 repeats; lane 2, 3 repeats; lane 3, 4 repeats; lane 4, 6 repeats; lane 5, 7 repeats; lane 6, 10 repeats; lane 7,...
Molecular weights of PCR-generated fragments for loci 1982 and 3232 for some isolates exceeded 1 kb, and results of automated fragment analysis were inconsistent. Molecular weights of PCR products and numbers of MIRU repeats for these strains were determined manually by electrophoresis on a 1.2% agarose gel (Agarose 1000, Invitrogen Ltd, Paisley, UK) with a 100-bp step DNA ladder as fragment size standard (Promega, Madison, WI, USA) (Figure).
Automated calling (in which PCR fragment sizes and allele assignment are automatically determined) or manual determination of genotyping data (number of repeats for each loci) was entered into Microsoft Excel (Redmond, WA, USA) tables and imported for further analysis into BioNumerics software (Applied Maths, St Martens, Belgium). Genetic distance analysis and cluster comparison were done by using a categorical variable index and the unweighted pair group method with arithmetic mean algorithm. If >2 isolates possessed identical VNTR signatures, they were considered to be clustered. For discrimination analysis, Hunter-Gaston diversity index (HGDI) was calculated as described (31) and used for comparison of the discriminatory power of VNTR typing for individual loci and for all loci taken together.
Baseline clinical and sociodemographic parameters of the 187 TB patients are shown in Table 1. MIRU-ETR analysis of the 187 isolates yielded 10 clusters of indistinguishable isolates and 58 unique patterns. The codes for the 15 loci were expressed in the following order: MIRU 2, 4, 10, 16, 20, 23, 24, 26, 27, 31, 39, 40, ETR-A, ETR-B, ETR-C. Cluster sizes varied from 2 to 75 isolates. The 2 largest clusters consisted of 31 strains with the 223325173533423 profile and 75 strains with the 223325153533423 MIRU-ETR profile, respectively, both consisting of Beijing strains (Table 2). For all strains, genotyping with the 15-MIRU–ETR loci set was more informative and had a higher discriminatory power than with spoligotyping, as expected (HGDI 0.747 for MIRU vs 0.572 for spoligotyping, data not shown).
The results of the allelic diversity analysis for all 187 isolates are summarized in Table 3. The discriminatory index for 5 loci (MIRU 26, MIRU 31, MIRU 39, MIRU 40, and ETR-A) exceeded 0.3; these loci were regarded as moderately discriminating according to definitions proposed in a recent study (23). Other loci were found to be less polymorphic, with HGDIs within the range 0 to 0.3; no polymorphism was registered for loci MIRU 24 and ETR-B. The maximal number of allelic variants (9) was registered for locus MIRU 10, although its discriminatory power was poor because of uneven distribution of isolates with different numbers of repeats.
Spoligotyping identified 129 isolates as belonging to the Beijing family (69.0%), a proportion similar to that previously reported for Samara Oblast (7,18). Most (123) of the Beijing isolates had the characteristic spoligotyping profile with 9 final spacers present in the DR region (35–43), whereas 6 isolates had incomplete profiles because they lacked spacer 40 (4 isolates) or 43 (2 isolates). Results of spoligotyping were verified by detection of the IS6110 insertion in the dnaA-dnaN region. All 129 isolates yielded a PCR-product ≈2,000 bp, indicative of Beijing strains. Isolates belonging to strains other then Beijing did not possess an insertion in this region and yielded an ≈550-bp PCR product.
The allelic diversity and HGDIs were calculated separately for Beijing strains (Table 4). These were subjected to VNTR analysis by using an additional panel of loci (0424, 0531, 1955, 1982, 2074, 2163a, 3232, 3239, 3336, and 3690; Table A1). The allelic diversity analysis results calculated by using 25-MIRU loci and discriminatory indices are presented in Table 5.
This study evaluated the discriminatory ability of VNTR analysis in a population of patients with TB, in which a predominant proportion of the infecting TB strains belonged to the Beijing family. This genetic group, along with the W family, have been recently shown to share similar characteristics, including absence of spacers 1–34 in the DR region of the M. tuberculosis genome (14,15,17). The latter characteristic, although well known and widely accepted, may not always be definitive for identification of Beijing/W strains alone, as spacers 35–43 are present in several other M. tuberculosis families. Moreover, some spacers (particularly 37, 38, and 40) are missing in certain Beijing/W isolates because of loss of the target for DRa and DRb primers caused by deletions or presence of IS6110 insertions in the DR region (32,33).
Results of spoligotyping were confirmed by detection of an IS6110 insertion between the dnaA and dnaN genes, which was found in all 129 Beijing isolates but not in the remaining isolates. Results of our analysis demonstrate 100% specificity of the method and its applicability for robust, rapid, and reliable identification of Beijing family strains.
In our study, 6 (4.7%) of 129 isolates identified as Beijing on the basis of spoligotyping had incomplete spoligotyping profiles with a missing spacer 40 or 43. As has been argued by Bifani et al. (32), W strains with missing spacer 40 are defined as W14 group, characterized by a specific IS6110 RFLP pattern, high levels of drug resistance, and an additional repeat in ETR-D (MIRU 4) locus, i.e., MIRU-ETR profile 23332515 (or 7) 3533423. However, we observed no differences in the number of repeats in the MIRU 4 locus for these 4 isolates. All 4 isolates were multidrug resistant strains. Of the 4 patients, all were male, 1 was HIV-infected, 2 were Russian (1 from Chechnya and 1 from Azerbaijan), and 2 had been vaccinated with BCG.
The data from the application of 15-MIRU–ETRs demonstrated the homogeneity and clonality of TB strains in this region of Russia: 135 (72.2%) of 187 strains were clustered into 10 groups, which indicates high rates of recent TB transmission in the Samara region. General trends for VNTR loci diversity and discriminatory power agreed (with some exceptions) with those reported previously (20,22,23,34). The overall allelic polymorphism and discriminatory power of the VNTR loci in this population were lower than that reported in previously published studies because of the large number of Beijing isolates in our test panel. This number reflects the actual prevalence of Beijing strains in Samara Oblast and in the few other regions in Russia for which the proportion of Beijing strains has been described (Table 2, Table 3, Table 4).
The 15-MIRU–ETR profiles 223325153533423 or 223325173533423 were shared by 106 (82.2%) of the total number of analyzed Beijing strains with an HGDI of 0.625 for all 15 loci. Four loci (MIRU20, MIRU24, MIRU27, and ETR-B) were monomorphic for Beijing strains, and the only locus with sufficient discriminatory power for differentiating among Beijing family strains was MIRU 26. This finding is in marked contrast to the application of MIRU-ETR in other patient populations in which the discriminatory power of MIRU, used in conjunction with spoligotyping, was arguably almost comparable to RFLP IS6110 (20–23,35). In these studies, HGDI values were subdivided into 3 groups, according to their ability to discriminate: poor (2, 20, 27), moderate (4, 16, 24, 39, ETR-B, ETR-C), and high (10, 23, 26, 31, 40, ETR-A) (23).
We expanded the panel of loci to improve discrimination among the Beijing isolates by using loci that had previously been reported to be highly polymorphic (24–27). The allelic diversity analysis and discriminatory indices are presented in Table 5.
The expanded set of VNTRs provided better discrimination than the original set of 15-MIRU–ETRs: 53 different profiles were identified, including 9 shared types in clusters (2–46 isolates each) and 44 unique patterns. The HGDI for 25 VNTR loci together was 0.870 (compared with 0.625 for the original set of MIRU-ETRs).
The most discriminatory individual VNTR were loci 3232 and 1982, with a large number of allelic variants (10 and 7, respectively) and HGDIs of 0.621 and 0.489, respectively. Other loci demonstrated poor discriminatory power (HGDI 0–0.2), and locus 2074 was monomorphic.
In our analysis of Russian Beijing isolates, 3 loci (MIRU 26, VNTR 1982, and VNTR 3232; 0.3–0.6) were sufficiently polymorphic for differentiation within the Beijing family. A relatively high degree of polymorphism in MIRU 26 has been previously reported (20,23,36). Loci 3232 and 1982 have been much less studied. Locus 3232 was shown to be more polymorphic in M. tuberculosis than in M. bovis (8 vs. 5 allelic variants, respectively) with moderate to high HGDI values (0.60). Locus 3232 displayed the highest discrimination power among 8 novel VNTR loci introduced by Roring et al. (24,25). The basis of polymorphism in loci MIRU 26, VNTR 3232, and VNTR 1982 is not completely clear. The first 2 loci are located in the intergenic regions of genes involved in metabolism of cell membrane components and transmembrane transport (locus VNTR 3232) and between genes Rv2679 (possible echA15, enoyl-CoA hydratase) and Rv 2680 (unknown protein) (locus MIRU 26). Locus VNTR 1982 is part of the Rv1753c (PPE24), which belongs to the group of genes encoding PPE proteins and which has been argued to be responsible for antigenic variation of M. tuberculosis (37). In our study, the degree of polymorphism at this locus was considerably higher than that previously reported for a panel of M. bovis isolates (27). By contrast, loci 2163a, 1955, 3336, and 3690, which were reported to be polymorphic for M. bovis, demonstrated low polymorphism in our study of M. tuberculosis Beijing isolates.
We speculate that the presence of variation at a small number of loci in genes likely to be involved in the earliest interactions of pathogen and host against a background of homogeneity at other loci suggests that these regions may be involved in the successful transmission of Beijing family strains. For the whole population studied, a small group of loci (VNTR 1982, VNTR 3232, MIRU 10, MIRU 26, MIRU 31, MIRU 39, MIRU 40, and ETR-A) offered the most discriminating panel and may be considered an essential part of the prospective universal panel of VNTR loci suitable for differentiation of M. tuberculosis isolates in different geographic settings with variable prevalence of highly conserved genotypes. The application of a limited number of loci (MIRU 10, MIRU 26, MIRU 31, VNTR 3232, and VNTR 1982) independently or in combination with other loci, may be a useful and rapid tool for differentiating strains within the Beijing family and for practical prospective genotyping and tracing of TB outbreaks in populations where the Beijing genotype predominates. Given the limited number of loci, analyzing this panel with a manual or automated approach is practical.
In conclusion, our study shows that by expanding the VNTR panel beyond the 15-MIRU–ETR loci described previously, discrimination can be substantially increased. This method may be used for typing M. tuberculosis isolates, even for populations in which a particular genetic group is dominant.
Dr Nikolayevskyy is a postdoctoral research assistant at the HPA Mycobacterium Reference Unit, Institute of Cell and Molecular Sciences, Barts and The London School of Medicine, University of London. His research interests include different aspects of the clinical microbiology, epidemiology, and molecular genetics of M. tuberculosis.
Acknowledgments
We thank the bacteriologists at the Samara Oblast TB Reference Laboratory and Samara Oblast Health authorities and TB service for their help in sample collection, processing, and organizational issues, and staff of the UK Health Protection Agency (HPA) Mycobacterium Reference Unit for their essential assistance and support.
This study was funded by UK Department for International Development (CNTR 000634) and a Foreign and Commonwealth Office Chevening Fellowship to V.N. (UKE0100129).
References
- Raviglione MC. The TB epidemic from 1992 to 2002. Tuberculosis (Edinb). 2003;83:4–14. DOIPubMedGoogle Scholar
- Yerokhin VV, Punga VV, Rybka LN. Tuberculosis in Russia and the problem of multiple drug resistance. Ann N Y Acad Sci. 2001;953:133–7. DOIPubMedGoogle Scholar
- Shilova MV. Specific features of the spread of tuberculosis in Russia at the end of the 20th century. Ann N Y Acad Sci. 2001;953:124–32. DOIPubMedGoogle Scholar
- Drobniewski FA, Balabanova YM. The diagnosis and management of multiple-drug-resistant tuberculosis at the beginning of the new millennium. Int J Infect Dis. 2002;6(Suppl.1):S21–31. DOIPubMedGoogle Scholar
- UNAIDS. 2004 Report of the global AIDS epidemic. Geneva: UNAIDS; 2004.
- Drobniewski FA, Atun R, Fedorin I, Bikov A, Coker R. The "bear trap": the colliding epidemics of tuberculosis and HIV in Russia. Int J STD AIDS. 2004;15:641–6. DOIPubMedGoogle Scholar
- Drobniewski F, Balabanova Y, Ruddy M, Weldon L, Jeltkova K, Brown T, Rifampin- and multidrug-resistant tuberculosis in Russian civilians and prison inmates: dominance of the Beijing strain family. Emerg Infect Dis. 2002;8:1320–6.PubMedGoogle Scholar
- Toungoussova OS, Sandven P, Mariandyshev AO, Nizovtseva NI, Bjune G, Cougant DA. Spread of drug-resistant Mycobacterium tuberculosis strains of the Beijing genotype in the Archangel oblast, Russia. J Clin Microbiol. 2002;40:1930–7. DOIPubMedGoogle Scholar
- van Soolingen D. Molecular epidemiology of tuberculosis and other mycobacterial infections: main methodologies and achievements. J Intern Med. 2001;249:1–26. DOIPubMedGoogle Scholar
- Braden CR, Crawford JT, Schable BA. Quality assessment of Mycobacterium tuberculosis genotyping in a large laboratory network. Emerg Infect Dis. 2002;8:1210–5.PubMedGoogle Scholar
- McHugh TD, Dickens A, Gillespie SH. False molecular clusters due to non-random association of IS6110 with Mycobacterium tuberculosis. J Clin Microbiol. 2000;38:2081–6.PubMedGoogle Scholar
- Kanduma E, McHugh TD, Gillespie SH. Molecular methods for Mycobacterium tuberculosis strain typing: a users guide. J Appl Microbiol. 2003;94:781–91. DOIPubMedGoogle Scholar
- Glynn JR, Whiteley J, Bifani PJ, Kremer K, van Soolingen D. Worldwide occurrence of Beijing/W strains of Mycobacterium tuberculosis: a systematic review. Emerg Infect Dis. 2002;8:843–9.PubMedGoogle Scholar
- Kremer K, Glynn JR, Lillebaek T, Niemann S, Kurepina N, Kreiswirth B, Definition of the Beijing/W Lineage of Mycobacterium tuberculosis on the Basis of Genetic Markers. J Clin Microbiol. 2004;42:4040–9. DOIPubMedGoogle Scholar
- Milan SJ, Hauge K, Kurepina N, Lofy K, Goldberg S, Narita M, Expanded geographical distribution of the N family of Mycobacterium tuberculosis strain within the United States. J Clin Microbiol. 2004;42:1064–8. DOIPubMedGoogle Scholar
- Sreevatsan S, Pan X, Stockbauer K, Connell N, Kreiswirth B, Whittam T, Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global transmission. Proc Natl Acad Sci U S A. 1997;94:9869–74. DOIPubMedGoogle Scholar
- Plikaytis BB, Marden J, Crawford J, Woodley C, Buter W, Shinnik T. Multiplex PCR assay specific for the multidrug-resistant strains W of Mycobacterium tuberculosis. J Clin Microbiol. 1994;32:1542–6.PubMedGoogle Scholar
- Drobniewski F, Balabanova Y, Nikolayevskyy V, Ruddy M, Kuznetzov S, Zakharova S, Drug-resistant TB, clinical virulence, and the dominance of the Beijing strain family in Russia. JAMA. 2005;293:2726–31. DOIPubMedGoogle Scholar
- Frothingham R, Meeker-O'Connell WA. Genetic diversity in the Mycobacterium tuberculosis complex based on variable numbers of tandem repeats. Microbiology. 1998;144:1189–96. DOIPubMedGoogle Scholar
- Cowan LS, Mosher L, Diem L, Massey JP, Crawford JT. Variable-number tandem repeats typing of Mycobacterium tuberculosis isolates with low copy numbers of IS6110 by using mycobacterial interspersed repetitive units. J Clin Microbiol. 2002;40:1592–602. DOIPubMedGoogle Scholar
- Supply P, Mazars E, Lesjean S, Vincent V, Gicquel B, Locht C. Variable human minisatellite-like regions in the Mycobacterium tuberculosis genome. Mol Microbiol. 2000;36:762–71. DOIPubMedGoogle Scholar
- Supply P, Lesjean S, Savine E, Kremer K, van Soolingen D, Locht C. Automated high-throughput genotyping for study of global epidemiology of Mycobacterium tuberculosis based on mycobacterial interspersed repetitive units. J Clin Microbiol. 2001;39:3563–71. DOIPubMedGoogle Scholar
- Sola C, Filliol I, Legrand E, Lesjean S, Locht C, Supply P, Genotyping of the Mycobacterium tuberculosis complex using MIRUs: association with VNTR and spoligotyping for molecular epidemiology and evolutionary genetics. Infect Genet Evol. 2003;3:125–33. DOIPubMedGoogle Scholar
- Roring S, Scott A, Brittain D, Walker I, Hewinson RG, Neill S, Development of variable-number tandem repeat typing of Mycobacterium bovis: comparison of results with those obtained by using existing exact tandem repeats and spoligotyping. J Clin Microbiol. 2002;40:2126–33. DOIPubMedGoogle Scholar
- Roring S, Scott AN, Hewinson RG, Neill SD, Skuce RA. Evaluation of variable number tandem repeat (VNTR) loci in molecular typing of Mycobacterium bovis isolates from Ireland. Vet Microbiol. 2004;101:65–73. DOIPubMedGoogle Scholar
- Le Flèche P, Fabre M, Denoeud F, Koeck J-L, Vergnaud G. High resolution, on-line identification of strains from the Mycobacterium tuberculosis complex based on tandem repeat typing. BMC Microbiol. 2002;2:37–48. DOIPubMedGoogle Scholar
- Skuce RA, McCorry TP, McCarroll JF, Roring SM, Scott AN, Brittain D, Discrimination of Mycobacterium tuberculosis complex bacteria using novel VNTR-PCR targets. Microbiology. 2002;148:519–28.PubMedGoogle Scholar
- Ruddy M, Balabanova Y, Graham C, Fedorin I, Malomanova N, Elisarova E, Rates of drug resistance and risk factor analysis in civilian and prison patients with tuberculosis in Samara Region, Russia. Thorax. 2005;60:130–5. DOIPubMedGoogle Scholar
- Yates MD, Drobniewski FA, Wilson SM. Evaluation of a rapid PCR-based epidemiological typing method for routine studies of Mycobacterium tuberculosis. J Clin Microbiol. 2002;40:712–4. DOIPubMedGoogle Scholar
- Kamerbeek J, Schouls L, Kolk A, van Agterveld M, van Soolingen D, Kuijper S, Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol. 1997;35:907–14.PubMedGoogle Scholar
- Hunter PR, Gaston MA. Numerical index of the discriminatory ability of typing systems: an application of Simpson's index of diversity. J Clin Microbiol. 1988;26:2465–6.PubMedGoogle Scholar
- Bifani P, Mathema B, Campo M, Moghazeh S, Nivin B, Shashkina E, Molecular identification of streptomycin monoresistant Mycobacterium tuberculosis related to multidrug resistant W strain. Emerg Infect Dis. 2001;7:842–8. DOIPubMedGoogle Scholar
- Mokrousov I, Narvskaya O, Limeschenko E, Otten T, Vyshnevskiy B. Novel IS6110 insertion sites in the direct repeat locus of Mycobacterium tuberculosis clinical strains from the St. Petersburg area of Russia and evolutionary and epidemiological considerations. J Clin Microbiol. 2002;40:1504–7. DOIPubMedGoogle Scholar
- Mokrousov I, Narvskaya O, Limeschenko E, Vyazovaya A, Otten T, Vyshnevskiy B. Analysis of the allelic diversity of the mycobacterial interspersed repetitive units in Mycobacterium tuberculosis strains of the Beijing family: practical implications and evolutionary considerations. J Clin Microbiol. 2004;42:2438–44. DOIPubMedGoogle Scholar
- Hawkey PM, Smith EG, Evans JT, Monk P, Bryan G, Mohamed HH, Mycobacterial interspersed repetitive unit typing of Mycobacterium tuberculosis compared to IS6110-based restriction fragment length polymorphism analysis for the investigation of apparently clustered cases of tuberculosis. J Clin Microbiol. 2003;41:3514–20. DOIPubMedGoogle Scholar
- Sun Y-J, Bellamy R, Lee AS, Ng ST, Ravindran S, Wong S-Y, Use of mycobacterial interspersed repetitive unit-variable-number tandem repeat typing to examine genetic diversity of Mycobacterium tuberculosis in Singapore. J Clin Microbiol. 2004;42:1986–93. DOIPubMedGoogle Scholar
- Cole ST, Brosch R, Parkhill J. 39 other authors. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–44. DOIPubMedGoogle Scholar
Figure
Tables
Cite This ArticleTable of Contents – Volume 12, Number 9—September 2006
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Francis Drobniewski, HPA Mycobacterium Reference Unit, Institute of Cell and Molecular Science, Barts and The London, Queen Mary’s School of Medicine, University of London, 2 Newark St, E1 2AT, London, UK
Top