Volume 12, Number 9—September 2006
Research
Predominance of Ancestral Lineages of Mycobacterium tuberculosis in India
Cite This Article
Citation for Media
Abstract
Although India has the highest prevalence of tuberculosis (TB) worldwide, the genetic diversity of Mycobacterium tuberculosis in India is largely unknown. A collection of 91 isolates originating from 12 different regions spread across the country were analyzed by genotyping using 21 loci with variable-number tandem repeats (VNTRs), by spoligotyping, by principal genetic grouping (PGG), and by deletion analysis of M. tuberculosis–specific deletion region 1. The isolates showed highly diverse VNTR genotypes. Nevertheless, highly congruent groupings identified by using the 4 independent sets of markers permitted a clear definition of 3 prevalent PGG1 lineages, which corresponded to the "ancestral" East African–Indian, the Delhi, and the Beijing/W genogroups. A few isolates from PGG2 lineages and a single representative of the presumably most recent PGG3 were identified. These observations suggest a predominance of ancestral M. tuberculosis genotypes in the Indian subcontinent, which supports the hypothesis that India is an ancient endemic focus of TB.
Tuberculosis (TB) in humans has been described since ancient times. Mycobacterium tuberculosis, its main causative agent, is widely disseminated and is one of the most successful human pathogens today, with 2 billion persons infected. Most of the disease's effects are now concentrated in countries with few resources; India has the highest number of cases (1).
Because of the clonal structure of M. tuberculosis (2–4), comparative genotypic analyses from widespread geographic areas, such as the Indian subcontinent, or from different human populations can give unique insights into dissemination dynamics and evolutionary genetics of the pathogen (5,6). IS6110 restriction fragment length polymorphism–based fingerprinting (7) has been used to study the mycobacterial population structure from southern India, northern India, and the Delhi region (8–11). However, IS6110 fingerprinting is of limited use because a high proportion of M. tuberculosis strains have low copy numbers or are devoid of IS6110 in several regions of India (8,10). IS6110 typing also has a relative lack of portability, which hinders comparison between separate studies (12). Fingerprinting methods targeting polymorphic spacer sequences in the direct repeat (DR) region, including spoligotyping, have been used in some of these regions and in Bombay (13–15). However, when used alone, these methods considerably underestimate the clonal diversity (16). Because of these limitations, knowledge about the mycobacterial population structure in India remains incomplete.
More recently, molecular typing methods based on variable number tandem repeats (VNTRs) of genetic elements named mycobacterial interspersed repetitive units (MIRUs) (17) have been developed (18,19). MIRU-VNTR typing shows a discriminatory power close to that of IS6110 fingerprinting and is particularly efficient in distinguishing M. tuberculosis isolates with few IS6110 elements or none (19–21). MIRU-VNTRs are sufficiently stable to track epidemic strains (19,20,22).
We analyzed M. tuberculosis strain diversity in a sample of 91 isolates from 12 different regions, including northern, central, and southern India, by using a set of 21 VNTR loci, including the 12 MIRU-VNTR loci described previously (17,18) and 9 additional loci containing VNTRs of other interspersed genetic elements (23–25). All of these loci are collectively designated MIRU-VNTR loci in this study. Spoligotyping was used as a complementary technique because this procedure, albeit less discriminatory, is useful in identifying genotype families (16,26,27). In addition, single nucleotide polymorphism (SNP) genotyping on the katG and gyrA genes and genomic deletion analysis with M. tuberculosis–specific deletion region 1 (TbD1) were used to assess consistency of the genetic relationships obtained by VNTR typing and spoligotyping at a broader evolutionary level. SNPs in the katG and gyrA genes classify M. tuberculosis isolates into 3 principal genetic groups (PGGs) thought to have evolved sequentially from group 1 to group 3 (2). TbD1 is specifically present in a subset of PGG1 strains, but absent in other strains of PGG1, and in PGG2 and PGG3 strains; TbD1+ strains have therefore been proposed to constitute an ancestral lineage of M. tuberculosis (28). Using a combination of all 4 markers, we found that ancestral lineages prevail in our collection, which suggests an ancient focus of TB in the Indian subcontinent.
Strains and Genomic DNA Extraction
A sample of 100 clinical isolates of M. tuberculosis was initially selected; the isolates originated in 12 different regions, from northern, central, and southern India and part of eastern India (Table 1). For 9 isolates, mixed infections or laboratory cross-contamination was suspected after MIRU-VNTR typing (see Results), and they were excluded from further analysis. The isolates were collected from patients with pulmonary TB who had voluntarily visited their nearest medical college or hospital for diagnosis and treatment. Therefore, in most of the cases, the patients lived near the respective cities reported in Table 1. Patients were adults, 20 to 45 years of age, and represented both men and women, except those from Ranchi where all reported cases were in male army personnel. Information regarding the extent of disease and treatment status (new or recurrent cases of disease) was not available. From these hospitals, most of the isolates (designated hereafter as ICC, VA, VK, HA, BC, and ASN) were transported to the repository collection maintained at the Jalma Institute in Agra for further characterization and drug susceptibility testing, which was performed by the proportion method. Table 2 includes sensitivity profiles of isolates tested at the Jalma laboratory. The isolates from New Delhi were collected between March 1997 and March 1998, from Ahmedabad between November 1996 and July 1997, from Ranchi between February and March 1999, from Chandigarh in September 1997, from Bangalore in November 1996, from Jammu between May and July 2001, from Jaipur in May 2001, from Agra in May 2001, and from Varanasi between June and November 1999. The isolates designated as MHRC were from pulmonary TB patients who sought treatment at the Mahavir Hospital, Hyderabad, between 2000 and 2002. The isolates designated as TRC were from pulmonary TB patients at the Tuberculosis Research Centre, Chennai. M. tuberculosis DNA was extracted by using the standardized protocol as described (7).
TbD1 Analysis
The presence of TbD1 was analyzed by PCR (28). Briefly, 2 PCR assays were performed per isolate tested, by using either primers complementary to the sequences flanking the deleted region or primers complementary to the internal sequences. For the isolates that did (TbD1+) or did not (TbD1–) contain the TbD1 region, an amplicon was obtained only with internal primers or only with flanking primers, respectively.
Single Nucleotide Polymorphism Analysis
To define the PGGs, the polymorphisms at the katG codon 463 and the gyrA codon 95 were determined by sequence analysis after PCR amplification with the same primers as in Sreevatsan et al. (2). The amplification products were sequenced by using an ABI 3700 DNA sequencer and the BigDye Terminator v3.1 Cycle sequencing kit (PE Applied Biosystems, Foster City, CA, USA).
Spoligotyping
Spoligotyping was performed by using a commercial kit (Isogen Bioscience BV, Maarsen, the Netherlands) according to the previously described method (29). Reverse blotting analysis of spacer sequences in the DR region was performed by using a streptavidin-horseradish peroxidase–enhanced enzyme chemiluminescence assay (Amersham Pharmacia-Biotech, Roosendaal, the Netherlands).
MIRU-VNTR Typing
The M. tuberculosis isolates were genotyped by PCR amplification of the 12 MIRU-VNTR loci (18) and 9 additional VNTR loci (23–25) by using an automated technique as described (19). The set of loci thus included (alternative designation in parentheses) MIRU-VNTR loci 2, 4 (ETR-D), 10, 16, 20, 23, 24, 26, 27, 31 (ETR-E), 39 and 40; and VNTR loci 424, 577 (ETR-C), 1895 (QUB-1895), 2347, 2401, 2461 (ETR-B), 3171, 3690, and 4156 (QUB-4156). The primers against the MIRU-VNTR flanking regions were the same as described (18), except that hex labeling was replaced by Vic labeling. The primers corresponding to the 9 additional VNTR loci and the conditions for their amplification by multiplex PCR are described in Table 3. The PCR fragments were sized and the various VNTR alleles were assigned after electrophoresis on a 96-well ABI 377 sequencer with customized GeneScan and Genotyper software packages (PE Applied Biosystems), as described (19) and on the basis of data described in Table 3. Tables used for MIRU-VNTR allele scoring are available at http://www.ibl.fr/mirus/mirus.html.
Analysis of Genetic Relationships
MIRU-VNTR profiles and spoligotypes were computed as character data into the Bionumerics program (Bionumerics version 2.5, Applied Maths, Saint-Martens-Latem, Belgium). MIRU-VNTR profiles were compared to each other by using the neighbor-joining algorithm. For spoligotypes, the Jaccard index was calculated to allow for the construction of a dendrogram by using the unweighted pair-group method with arithmetic averages. The spoligotypes were compared to fingerprints in an international database (31) that contained fingerprints from 13,008 isolates at the time of the consultation (June 2004). The genetic relationships between the isolates based on the MIRU-VNTR types were assessed by matching the spoligotypes with TbD1, and SNP analyses were carried out on a selected set of isolates representative of the different spoligotypes in each of the 3 predicted PGG1 groups and in the predicted PGG2/3 groups.
MIRU-VNTR and Spoligotype Analysis
Nine of the 100 isolates of the study collection displayed 2 alleles in several independent MIRU-VNTR loci among the 21 tested, which suggested mixed DNA populations. These mixed populations could have originated from laboratory cross-contaminations or from mixed infections. Therefore, they were excluded from further analysis.
Figure 1

Figure 1. Genetic relationships of Indian Mycobacterium tuberculosis isolates. The dendrogram based on mycobacterial interspersed repetitive units–variable-number tandem repeats (MIRU-VNTR) genotypes, generated by using the neighbor-joining algorithm, was rooted with M. canettii, the...
The remaining 91 isolates showed highly diverse MIRU-VNTR genotypes (Figure 1). Seventy-eight distinct genotypes were detected in this collection, including 6 cluster patterns and 72 unique patterns. The largest MIRU-VNTR cluster included 8 isolates, 5 of which originated in Ranchi. Another cluster included 3 isolates from New Delhi, while the remaining 4 clusters contained 2 isolates each (1 with 2 isolates from Delhi; the others included isolates from Jammu and Chandigarh, from Hyderabad and Chennai, and from Bangalore and Chandigarh). Information about possible links between patients with clustered isolates was not available. The number of different spoligotypes (36 distinct spoligotypes, including 11 cluster patterns and 25 unique patterns) was lower than that of the MIRU-VNTR types, which was consistent with previous comparisons between spoligotyping and MIRU-VNTR systems based on 12 loci (19–21). None of the MIRU-VNTR clusters was split by spoligotyping, while of the 11 spoligotype clusters, 9 were split by MIRU-VNTR typing.
The genetic relationships between the isolates based on the MIRU-VNTR types by using the neighbor-joining algorithm are displayed in Figure 1. This dendrogram indicates 3 main genotype groups. The identity of these groups was inferred by comparison with genetically well-characterized isolates from a worldwide collection (16), typed by using the same 21 MIRU-VNTR loci (19; P. Supply et al., unpub. data). These groups are thereby predicted to correspond to the TbD1+ ancestral lineage (41 isolates, 45% of the total isolates), the recently described Delhi or Central Asian (CAS) genogroup (24 isolates, 26%), and the Beijing genogroup (9 isolates, 10%), respectively. These 3 groups belong to PGG1. The remaining isolates (17 isolates, 19%) are predicted to belong to PGG2 or PGG3 genogroups.
Congruence of Groupings Between Markers
In accordance with the MIRU-VNTR typing results and the absence or presence of DR spacers 33–36 (26), katG and gyrA sequence analyses identified all tested representatives from Delhi and the ancestral genogroups as PGG1, whereas 14 tested isolates were assigned to PGG2 (Figure 1). One representative of PGG3, assumed to be the most recent group, was detected in this sample. The Beijing/W isolates were not tested for katG and gyrA polymorphism, since the fact that they all belong to PGG1 is well documented (26).
In agreement with Brosch et al. (28), we found the TbD1 region in all tested isolates from the predicted ancestral group but not in all tested Beijing/W, PGG2, and PGG3 isolates (Figure 1). We also found that all the tested Delhi isolates lacked TbD1.
Already known spoligotype signatures (16,26–28,31,32), and a few new variants, were found within the 4 groups defined by MIRU-VNTR analysis (Figure 1 and Table 4). The TbD1+ isolates were characterized by the absence of spacers 29 to 32 and 34, and (except in 4 cases) by the presence of spacer 33. Most isolates (35 of 43, taking into account 2 Indian isolates from the collection of Kremer et al. [16]) also lacked spacers 2 and 3. Based on these results, three fourths of the TbD1+ isolates were included in the spoligotype EAI3 class (33), while 1 isolate belonged to the EAI1 class. The remaining TbD1+ isolates represented new EAI variants. EAI classes 2, 4, and 5 were not found in this collection. Typically, the Beijing/W isolates only harbored spacers 35–43 (with spacers 39 and 40 missing in 1 case) (5,32). As described recently (11), the Delhi isolates shared the block of 9 final spacers (with some internal variation) with the Beijing/W strains but included 2 additional blocks among spacers 1–22. They specifically lacked spacers 4–7, and 23–34. The Delhi types are thus included in the CAS spoligotype family (33). Fourteen and 4 isolates out of 25 (taking into account 1 Indian isolate from the collection of Kremer et al. [16]) conformed to the 2 main spoligotype prototypes, CAS1 and CAS2, respectively (33).
As expected, the prototypes of the Latin American–Mediterranean (LAM, 3 cases), X (1 case), and T (8 cases) spoligotype families were detected among the isolates of the PGG2/3group. The single PGG3 isolate (ICC399) had a T spoligotype, which includes both PGG2 and PGG3 strains (26,33). Highly similar groupings of the isolates were observed when a dendrogram was built based on spoligotypes alone or on a combination of VNTR and spoligotypes, although the resolution was lower when spoligotyping was used alone (data not shown).
Comparison of Spoligotypes with an International Database
Table 5 shows the results from a comparison of the Indian spoligotypes with SpolDB3.0, a database containing data from >13,000 M. tuberculosis complex isolates obtained worldwide (31). Of 36 different spoligotypes found in the Indian strains, 15 (41.7%) were not present in SpolDB3.0. Most (11, 73%) of these new spoligotypes correspond to PGG1 isolates. Conversely, only 1 Indian isolate had the second most frequent spoligotype worldwide, S53. These observations reflect the current underrepresentation of strains from India in SpolDB3.0 (n = 44).
This report describes the diversity of M. tuberculosis strains obtained from patients in various regions in India, relying on a conveniently available set of isolates collected between 1997 and 2002. While these data are not representative of all TB patients in those regions and lack information regarding clinical characteristics, they provide valuable first insights into the diversity of circulating M. tuberculosis strains in this country. The excellent congruence observed between the 4 independent sets of genetic markers used here lends strong support to the assignment of different prevalent lineages. This congruence is consistent with the clonal population structure of M. tuberculosis (2–4) and reflects the respective informative values of the markers used. In particular, the results show that the use of a large set of VNTR loci simultaneously allows for both reliable identification of genogroups and high-resolution analysis of intralineage diversity, without the limitations that apply to IS6110 fingerprinting or other typing methods used in the few previous molecular studies on Indian isolates. Within the framework of the current evolutionary scenario of M. tuberculosis, which proposes phylogenies based on PGGs and genomic deletion analyses (e.g., TbD1) (2,3,28), we found a striking prevalence of ancestral genotypes (TbD1+) and the concurrent poor representation of the most recent lineages in this Indian collection (PGG2 and especially PGG3). This finding contrasts with the situation in other regions of the world, such as Europe and North and South America, where PGG2 and PGG3 constitute most of the M. tuberculosis strains (31).
Ancestral isolates of M. tuberculosis are characterized by the presence of the TbD1 region, which has been recently identified as an evolutionary landmark in the genome of this species. This region was detected initially in a few M. tuberculosis strains belonging to PGG1, as well as in M. canettii, M. bovis, M. africanum, and M. microti, whereas this region was shown to be absent in all PGG2 and PGG3 strains as well as in the other PGG1 strains tested (28). The grouping of all the tested TbD1+ isolates by MIRU-VNTR typing and spoligotyping (16,19, this study) support their assignment to a single lineage (28), the East African–Indian lineage (27). Consistently, all tested representatives of known modern M. tuberculosis genotype families were TbD1–. A similar systematic association has recently been observed in strains from Singapore (34) and from Bangladesh (35), which supports the notion that the deletion of TbD1 occurred as a single evolutionary event in a common ancestor rather than on independent multiple occasions (28).
In this study, all isolates that contain >2 repeats in MIRU-VNTR locus 24 belong to the ancestral (TbD1+ group, and all but 2 isolates containing 1 repeat unit in locus 24 belong to the modern (TbD1–) groups. This correlation, also seen in previous studies on isolates from Singapore (34) and Bangladesh (35), indicates that this locus alone is highly informative in the identification of ancestral and modern M. tuberculosis strains.
Figure 2
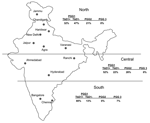
Figure 2. Geographic distribution of Mycobacterium. tuberculosis genotypes in northern, central, and southern India. PGG, principal genetic group; TbD1, M. tuberculosis–specific deletion region 1.
The few previously identified TbD1+ strains were isolated from patients from East Africa and South Asia. These strains have low copy numbers of IS6110 (16,28) and belong to cluster I within PGG1 (3). This lineage is distinct from IS6110 low-copy-number strains in PGG2 (3), which was isolated from patients in English-speaking countries (33). IS6110 low- copy-number strains are prevalent with variable proportions among patients from several countries in Southeast Asia (36), and the analysis of the available spoligotype data suggests that most of them belong to the TbD1+/EAI lineage (26,31,37). Frequencies of TbD1+/EAI isolates have recently been reported to range from 25% to 50% in Bangladesh (35,36) and Singapore (34). A frequency of 8% has been reported in a study that only used spoligotyping for genetic characterization of 105 isolates from the Delhi area (15). Until now, the highest prevalence of IS6110 low-copy-number isolates (≈60%) has been observed in southern India (8,9,38). Consistent with these studies, we found that 80% of the samples obtained from the southern regions from India were TbD1+/EAI isolates, although such isolates were found in nearly all regions (Figure 2). Also consistent with our findings, most spoligotypes observed in an ongoing population-based study (>1,200 isolates) in the southern state of Tamil Nadu were of the EAI3 class (S. Narayanan et al., unpub. data), found to be predominant in our collection (Table 4 and Figure 1).
The prevalence of these low-copy-number strains in regions of such high endemicity has raised the question of the true extent of genetic variation beyond their restricted IS6110 distribution (9). The MIRU-VNTR typing results obtained here indicate that the genetic diversity in the TbD1+/EAI lineage goes far beyond the commonly observed restricted spectrum of IS6110 low copy-number fingerprints and of known spoligotypes (31,36). For instance, most isolates with identical spoligotype 11 of the predominant EAI3 class in this study were of different MIRU-VNTR types (Figure 1). Moreover, the EAI lineage contains 3 additional spoligo-prototypes (EAI2, 4, and 5) that were absent from the population studied here. In addition, at least 1 group of TbD1+/EAI strains recently isolated in Singapore had high-copy numbers (up to 15) of IS6110 (39). Altogether these observations indicate that the TbD1+ strains constitute a highly diversified lineage, which is consistent with an ancestral phylogenetic position.
In addition to the TbD1+ isolates, 2 other major PGG1 families were well represented in this Indian collection. They were qualified as modern groups by their TbD1– status. The recently identified Delhi type (11), classified as the CAS group by spoligotyping (33), represented approximately one fourth of the total sample. This genogroup made up 60 (72%) of 83 isolates collected from male patients attending 1 hospital and a clinic of the Delhi region over a 1-year period (11) and 38 (36%) of 105 isolates collected from patients attending other health centers in Delhi (15). Although this genogroup is less dominant in this region, representing 5 (20%) of the 26 isolates from Delhi, the Delhi genogroup was well represented among the isolates from northern and central India as well. The second TbD1– PGG1 family detected in this study corresponds to the widespread Beijing/W family, which accounted for 10% of the total sample. Most (7 of 9) of these isolates were from Delhi, where they represented 30% of the isolates studied, in contrast to the 1% to 8% noted in other studies on isolates from Delhi or other Indian regions (11,15,32).
The predominance of M. tuberculosis ancestral strains and the relatively poor representation of the most recent lineages in this Indian collection lend support to the hypothesis that India is a relatively ancient endemic focus of TB (28). On the basis of these findings, we speculate that the Indian subcontinent was an early step of the worldwide expansion of the M. tuberculosis complex, subsequent to the recently proposed emergence of tubercle bacilli in eastern Africa millions of years ago (40). However, we acknowledge that, as our collection represents a minuscule fraction of the millions of TB cases in India, genotyping additional isolates from TB patients in this country will be necessary to determine if these initial observations hold true (as suggested by unpublished data from >1,200 isolates from southern Tamil Nadu) or substantially change for a larger fraction of reported cases. Nevertheless, we believe that our data constitute the most solid available foundation for future comparisons of these additional isolates and those obtained from patients in the rest of the world.
Dr Gutierrez is a senior scientist at the Reference Laboratory for Mycobacteria, Institut Pasteur. Her current research interests include the evolution and molecular epidemiology of the tubercle bacilli and other mycobacteria.
Acknowledgments
M. Hanif, V.K. Kataria, B. Malhotra, D. Khandalia, M. Sharma, and several research students from India are gratefully thanked for providing isolates.
This work was supported by a joint grant from the Institut National de la Santé et de la Recherche Médicale and the Indian Council for Medical Research.
References
- World Health Organization. Global tuberculosis control: surveillance, planning, financing (WH/HTM/TB/2004.331). Geneva: The Organization; 2004.
- Sreevatsan S, Pan X, Stockbauer KE, Connell ND, Kreiswirth BN, Whittam TS, Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination. Proc Natl Acad Sci U S A. 1997;94:9869–74. DOIPubMedGoogle Scholar
- Gutacker MM, Smoot JC, Migliaccio CA, Ricklefs SM, Hua S, Cousins DV, Genome-wide analysis of synonymous single nucleotide polymorphisms in Mycobacterium tuberculosis complex organisms: resolution of genetic relationships among closely related microbial strains. Genetics. 2002;162:1533–43.PubMedGoogle Scholar
- Supply P, Warren RM, Banuls AL, Lesjean S, Van Der Spuy GD, Lewis LA, Linkage disequilibrium between minisatellite loci supports clonal evolution of Mycobacterium tuberculosis in a high tuberculosis incidence area. Mol Microbiol. 2003;47:529–38. DOIPubMedGoogle Scholar
- Bifani PJ, Mathema B, Kurepina NE, Kreiswirth BN. Global dissemination of the Mycobacterium tuberculosis W-Beijing family strains. Trends Microbiol. 2002;10:45–52. DOIPubMedGoogle Scholar
- Hirsh AE, Tsolaki AG, DeRiemer K, Feldman MW, Small PM. Stable association between strains of Mycobacterium tuberculosis and their human host populations. Proc Natl Acad Sci U S A. 2004;101:4871–6. Epub 2004 Mar 23. DOIPubMedGoogle Scholar
- van Embden JD, Cave MD, Crawford JT, Dale JW, Eisenach KD, Gicquel B, Strain identification of Mycobacterium tuberculosis by DNA fingerprinting: recommendations for a standardized methodology. J Clin Microbiol. 1993;31:406–9.PubMedGoogle Scholar
- Das S, Paramasivan CN, Lowrie DB, Prabhakar R, Narayanan PR. IS6110 restriction fragment length polymorphism typing of clinical isolates of Mycobacterium tuberculosis from patients with pulmonary tuberculosis in Madras, south India. Tuber Lung Dis. 1995;76:550–4. DOIPubMedGoogle Scholar
- Radhakrishnan I, K MY, Kumar RA, Mundayoor S, Harris KA, Jr., Mukundan U, Implications of low frequency of IS6110 in fingerprinting field isolates of Mycobacterium tuberculosis from Kerala, India. J Clin Microbiol. 2001;39:1683. DOIPubMedGoogle Scholar
- Siddiqi N, Shamim M, Amin A, Chauhan DS, Das R, Srivastava K, Typing of drug resistant isolates of Mycobacterium tuberculosis from India using the IS6110 element reveals substantive polymorphism. Infect Genet Evol. 2001;1:109–16. DOIPubMedGoogle Scholar
- Bhanu NV, van Soolingen D, van Embden JD, Dar L, Pandey RM, Seth P. Predominance of a novel Mycobacterium tuberculosis genotype in the Delhi region of India. Tuberculosis (Edinb). 2002;82:105–12. DOIPubMedGoogle Scholar
- Braden CR, Crawford JT, Schable BA. Quality assessment of Mycobacterium tuberculosis genotyping in a large laboratory network. Emerg Infect Dis. 2002;8:1210–5.PubMedGoogle Scholar
- Narayanan S, Sahadevan R, Narayanan PR, Krishnamurthy PV, Paramasivan CN, Prabhakar R. Restriction fragment length polymorphism of Mycobacterium tuberculosis strains from various regions of India, using direct repeat probe. Indian J Med Res. 1997;106:447–54.PubMedGoogle Scholar
- Mistry NF, Iyer AM, D'Souza DT, Taylor GM, Young DB, Antia NH. Spoligotyping of Mycobacterium tuberculosis isolates from multiple-drug-resistant tuberculosis patients from Bombay, India. J Clin Microbiol. 2002;40:2677–80. DOIPubMedGoogle Scholar
- Singh UB, Suresh N, Bhanu NV, Arora J, Pant H, Sinha S, Predominant tuberculosis spoligotypes, Delhi, India. Emerg Infect Dis. 2004;10:1138–42.PubMedGoogle Scholar
- Kremer K, van Soolingen D, Frothingham R, Haas WH, Hermans PW, Martin C, Comparison of methods based on different molecular epidemiological markers for typing of Mycobacterium tuberculosis complex strains: interlaboratory study of discriminatory power and reproducibility. J Clin Microbiol. 1999;37:2607–18.PubMedGoogle Scholar
- Supply P, Magdalena J, Himpens S, Locht C. Identification of novel intergenic repetitive units in a mycobacterial two-component system operon. Mol Microbiol. 1997;26:991–1003. DOIPubMedGoogle Scholar
- Supply P, Mazars E, Lesjean S, Vincent V, Gicquel B, Locht C. Variable human minisatellite-like regions in the Mycobacterium tuberculosis genome. Mol Microbiol. 2000;36:762–71. DOIPubMedGoogle Scholar
- Supply P, Lesjean S, Savine E, Kremer K, van Soolingen D, Locht C. Automated high-throughput genotyping for study of global epidemiology of Mycobacterium tuberculosis based on mycobacterial interspersed repetitive units. J Clin Microbiol. 2001;39:3563–71. DOIPubMedGoogle Scholar
- Mazars E, Lesjean S, Banuls AL, Gilbert M, Vincent V, Gicquel B, High-resolution minisatellite-based typing as a portable approach to global analysis of Mycobacterium tuberculosis molecular epidemiology. Proc Natl Acad Sci U S A. 2001;98:1901–6. DOIPubMedGoogle Scholar
- Cowan LS, Mosher L, Diem L, Massey JP, Crawford JT. Variable-number tandem repeat typing of Mycobacterium tuberculosis isolates with low copy numbers of IS6110 by using mycobacterial interspersed repetitive units. J Clin Microbiol. 2002;40:1592–602. DOIPubMedGoogle Scholar
- Savine E, Warren RM, van der Spuy GD, Beyers N, van Helden PD, Locht C, Stability of variable-number tandem repeats of mycobacterial interspersed repetitive units from 12 loci in serial isolates of Mycobacterium tuberculosis. J Clin Microbiol. 2002;40:4561–6. DOIPubMedGoogle Scholar
- Frothingham R, Meeker-O'Connell WA. Genetic diversity in the Mycobacterium tuberculosis complex based on variable numbers of tandem DNA repeats. Microbiology. 1998;144:1189–96. DOIPubMedGoogle Scholar
- Roring S, Scott A, Brittain D, Walker I, Hewinson G, Neill S, Development of variable-number tandem repeat typing of Mycobacterium bovis: comparison of results with those obtained by using existing exact tandem repeats and spoligotyping. J Clin Microbiol. 2002;40:2126–33. DOIPubMedGoogle Scholar
- Le Fleche P, Fabre M, Denoeud F, Koeck JL, Vergnaud G. High resolution, on-line identification of strains from the Mycobacterium tuberculosis complex based on tandem repeat typing. BMC Microbiol. 2002;2:37. DOIPubMedGoogle Scholar
- Soini H, Pan X, Amin A, Graviss EA, Siddiqui A, Musser JM. Characterization of Mycobacterium tuberculosis isolates from patients in Houston, Texas, by spoligotyping. J Clin Microbiol. 2000;38:669–76.PubMedGoogle Scholar
- Sola C, Filliol I, Legrand E, Mokrousov I, Rastogi N. Mycobacterium tuberculosis phylogeny reconstruction based on combined numerical analysis with IS1081, IS6110, VNTR, and DR-based spoligotyping suggests the existence of two new phylogeographical clades. J Mol Evol. 2001;53:680–9. DOIPubMedGoogle Scholar
- Brosch R, Gordon SV, Marmiesse M, Brodin P, Buchrieser C, Eiglmeier K, A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci U S A. 2002;99:3684–9. DOIPubMedGoogle Scholar
- Kamerbeek J, Schouls L, Kolk A, van Agterveld M, van Soolingen D, Kuijper S, Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol. 1997;35:907–14.PubMedGoogle Scholar
- Warren RM, Victor TC, Streicher EM, Richardson M, van der Spuy GD, Johnson R, Clonal expansion of a globally disseminated lineage of Mycobacterium tuberculosis with low IS6110 copy numbers. J Clin Microbiol. 2004;42:5774–82. DOIPubMedGoogle Scholar
- Filliol I, Driscoll JR, van Soolingen D, Kreiswirth BN, Kremer K, Valetudie G, Snapshot of moving and expanding clones of Mycobacterium tuberculosis and their global distribution assessed by spoligotyping in an international study. J Clin Microbiol. 2003;41:1963–70. DOIPubMedGoogle Scholar
- van Soolingen D, Qian L, de Haas PE, Douglas JT, Traore H, Portaels F, Predominance of a single genotype of Mycobacterium tuberculosis in countries of east Asia. J Clin Microbiol. 1995;33:3234–8.PubMedGoogle Scholar
- Filliol I, Driscoll JR, Van Soolingen D, Kreiswirth BN, Kremer K, Valetudie G, Global distribution of Mycobacterium tuberculosis spoligotypes. Emerg Infect Dis. 2002;8:1347–9.PubMedGoogle Scholar
- Sun YJ, Bellamy R, Lee AS, Ng ST, Ravindran S, Wong SY, Use of mycobacterial interspersed repetitive unit-variable-number tandem repeat typing to examine genetic diversity of Mycobacterium tuberculosis in Singapore. J Clin Microbiol. 2004;42:1986–93. DOIPubMedGoogle Scholar
- Banu S, Gordon SV, Palmer S, Islam R, Ahmed S, Alam KM, Genotypic analysis of Mycobacterium tuberculosis in Bangladesh and prevalence of the Beijing strain. J Clin Microbiol. 2004;42:674–82. DOIPubMedGoogle Scholar
- Shamputa IC, Rigouts L, Eyongeta LA, El Aila NA, van Deun A, Salim AH, Genotypic and phenotypic heterogeneity among Mycobacterium tuberculosis isolates from pulmonary tuberculosis patients. J Clin Microbiol. 2004;42:5528–36. DOIPubMedGoogle Scholar
- Dale JW, Al-Ghusein H, Al-Hashmi S, Butcher P, Dickens AL, Drobniewski F, Evolutionary relationships among strains of Mycobacterium tuberculosis with few copies of IS6110. J Bacteriol. 2003;185:2555–62. DOIPubMedGoogle Scholar
- Narayanan S, Das S, Garg R, Hari L, Rao VB, Frieden TR, Molecular epidemiology of tuberculosis in a rural area of high prevalence in South India: implications for disease control and prevention. J Clin Microbiol. 2002;40:4785–8. DOIPubMedGoogle Scholar
- Sun YJ, Lee AS, Ng ST, Ravindran S, Kremer K, Bellamy R, Characterization of ancestral Mycobacterium tuberculosis by multiple genetic markers and proposal of genotyping strategy. J Clin Microbiol. 2004;42:5058–64. DOIPubMedGoogle Scholar
- Gutierrez MC, Brisse S, Brosch R, Omais B, Marmiesse M, Supply P, Ancient origin and gene mosaicism of tubercle bacilli. PLoS Pathog. 2005;1:e5. DOIPubMedGoogle Scholar
Figures
Tables
Cite This Article1These authors contributed equally to this article.
Table of Contents – Volume 12, Number 9—September 2006
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Philip Supply, INSERM U629, Institut Pasteur de Lille, 1 Rue du Prof Calmette, 59019 Lille CEDEX, France
Top