Volume 14, Number 11—November 2008
Dispatch
Change in Japanese Encephalitis Virus Distribution,Thailand
Figure 2
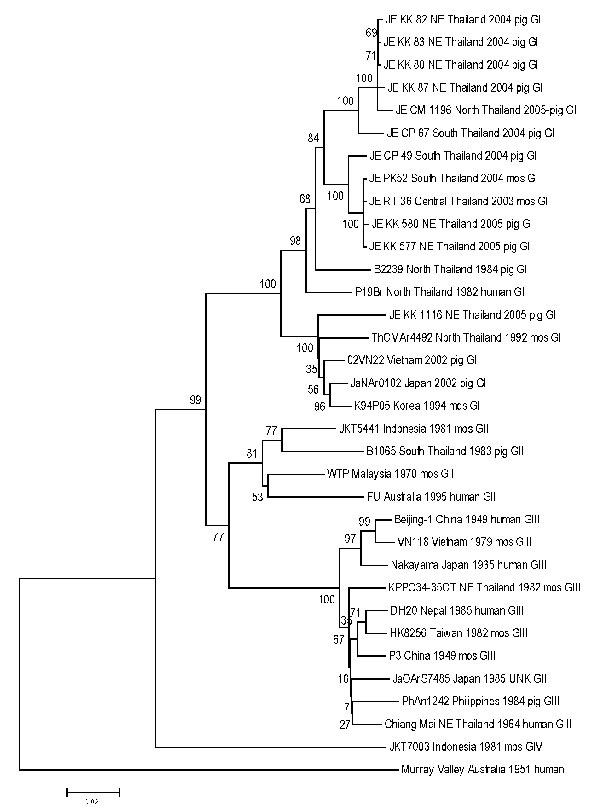
Figure 2. Sequence phylogeny based on E (envelope) gene nucleotide sequence of Japanese encephalitis virus isolates from pigs and mosquito hosts in Thailand during 2003–2005, with reference to other Southeast Asian isolates. Phylogenetic analysis was performed by using nucleotide alignments, the Kimura 2-parameter algorithm (for the calculation of pairwise distances), and the neighbor-joining method (for tree reconstruction), as implemented in MEGA software (9). The tree was rooted within the Japanese encephalitis serogoup by using Murray Valley virus (GenBank accession nos. E1–51). The robustness of branching patterns was tested by 1,000 bootstrap pseudoreplications. Each strain is abbreviated, followed by the country of origin (and the region of origin in Thailand, e.g., NE = northeast) and year of isolation. Bootstrap values are indicated above the major branch; 33 taxa comprised the ingroup, and all taxa were rooted with Murray Valley virus. A unique gap was treated as a "fifth base." The character state optimization was chosen as accelerated transformation. Consistence index 0.572; retention index 0.7528.
References
- Solomon T, Dung NM, Kneen M, Gainsborough M, Vaughn DW, Khanh VT. Japanese encephalitis. J Neurol Neurosurg Psychiatry. 2000;68:405–15. DOIPubMedGoogle Scholar
- Pyke AT, Williams DT, Nisbet DJ, van den Hurk AF, Taylor CT, Johansen CA, The appearance of a second genotype of Japanese encephalitis virus in the Australasian region. Am J Trop Med Hyg. 2001;65:747–53.PubMedGoogle Scholar
- Tsai TR, Solomon T, Vaughn DW. Flaviviruses (yellow fever, dengue, dengue hemorrhagic fever, Japanese encephalitis, West Nile, St Louis encephalitis, tick-borne encephalitis). In: Mandell GL, Bennett JE, Dolin R, editors. Principles and practice of infectious diseases, 6th ed. Philadelphia: Elsevier; 2004. p. 1926–50.
- Vaughn DW, Hoke CH Jr. The epidemiology of Japanese encephalitis: prospects for prevention. Epidemiol Rev. 1992;14:197–221.PubMedGoogle Scholar
- Solomon T, Ni H, Beasley DW, Ekkelenkamp M, Cardosa MJ, Barrett AD. Origin and evolution of Japanese encephalitis virus in Southeast Asia. J Virol. 2003;77:3091–8. DOIPubMedGoogle Scholar
- Solomon T. New vaccines for Japanese encephalitis. Lancet Neurol. 2008;7:116–8. DOIPubMedGoogle Scholar
- Chunsuttiwat S. Japanese encephalitis in Thailand. Southeast Asian J Trop Med Public Health. 1989;20:593–7.PubMedGoogle Scholar
- Moureau G, Temmam S, Gonzalez JP, Charrel RN, Grard G, de Lamballerie X. A real-time RT-PCR method for the universal detection and identification of flaviviruses. Vector Borne Zoonotic Dis. 2007;7:467–77. DOIPubMedGoogle Scholar
- Kumar S, Tamura K, Jakobsen IB, Nei M. MEGA2: Molecular Evolutionary Genetics Analysis software. Bioinformatics. 2001;17:1244–5. DOIPubMedGoogle Scholar
- Jan LR, Yueh YY, Wu YC, Horng CB, Wang GR. Genetic variation of Japanese encephalitis virus in Taiwan. Am J Trop. 2000;62:446–52.
- Mackenzie JS, Gubler DJ, Petersen LR. Emerging flaviviruses: the spread and resurgence of Japanese encephalitis, West Nile and dengue viruses. Nat Med. 2004;10(Suppl):S98–109. DOIPubMedGoogle Scholar
- van den Hurk AF, Johansen CA, Zborowski P, Paru R, Foley PN, Beebe NW, Mosquito host-feeding patterns and implications for Japanese encephalitis virus transmission in northern Australia and Papua New Guinea. Med Vet Entomol. 2003;17:403–11. DOIPubMedGoogle Scholar
- Ferguson M, Johnes S, Li L, Heath A, Barrett A. Effect of genomic variation in the challenge virus on the neutralization titres of recipients of inactivated JE vaccines—report of a collaborative study on PRNT50 assays for Japanese encephalitis virus (JE) antibodies. Biologicals. 2008;36:111–6. DOIPubMedGoogle Scholar
- Nitatpattana N, Apiwathnasorn C, Barbazan P, Leemingsawat S, Yoksan S, Gonzalez JP. First isolation of Japanese encephalitis from Culex quinquefasciatus in Thailand. Southeast Asian J Trop Med Public Health. 2005;36:875–8.PubMedGoogle Scholar