Volume 15, Number 12—December 2009
Dispatch
Molecular Epidemiology of Glanders, Pakistan
Cite This Article
Citation for Media
Abstract
We collected epidemiologic and molecular data from Burkholderia mallei isolates from equines in Punjab, Pakistan from 1999 through 2007. We show that recent outbreaks are genetically distinct from available whole genome sequences and that these genotypes are persistent and ubiquitous in Punjab, probably due to human-mediated movement of equines.
Glanders is an equine disease that was recognized by Hippocrates and Aristotle (1). It is caused by the bacterium Burkholderia mallei, an obligate pathogen of horses, donkeys, and mules (Equidae), with occasional infections in felines, canines, and humans (2,3). Strict regulations of equines have reduced the range of this once globally distributed disease to a few endemic foci in South and Central America, the Middle East, and parts of Africa and Asia (2,4). This emerging disease has only recently regained attention following the listing of B. mallei as a Category B agent by the US Centers for Disease Control and Prevention (2). Although outbreaks are common in regions of disease endemicity, much of what is known about the ecology and natural population dynamics of B. mallei relies on indirect evidence and expert opinion, with little to no knowledge concerning its genetic diversity (2,3). We genetically characterized 15 samples of B. mallei from recent outbreaks in Pakistan to provide additional knowledge of how this disease of antiquity is transmitted throughout endemic regions today.
We obtained clinical samples and background information from 15 glanderous equids in Punjab, Pakistan from 1999 through 2007 (Table; Appendix Table). Research on equine subjects was approved by the Synopsis Scrutiny Committee and Animal Ethics Committee, Faculty of Veterinary Science, University of Agriculture, Faisalabad, Pakistan. Samples were plated on brain-heart infusion (BHI) agar with 4% glycerol and incubated for 24–30 hours at 37°C. Individual colonies were inoculated into BHI broth containing 4% glycerol and were incubated with shaking for 36 h at 37°C. An aliquot of broth (1.5 mL) was centrifuged at 13,000 rpm for 15 min. Genomic DNA was extracted from the resulting pellets using standard digestion buffer and phenol-chloroform extraction protocols (6).
For genotyping, we screened 23 loci (Appendix Table) from a previously established 32-marker multiple locus variable number of tandem repeats (VNTR) analysis system designed for B. pseudomallei and B. mallei (7). In silico genotyping of the same loci was also performed for 10 whole genome sequences (WGS) of B. mallei (8; Appendix Table). VNTR markers have higher mutation rates than other genetic markers which make them inappropriate for determining deep levels of evolutionary relatedness, however VNTRS are appropriate for 1) discrimination between closely related isolates, 2) determination of the degree of relatedness among isolates, and 3) discernment of population structure on a spatial scale (7,9,10). This utility is especially important for B. mallei because it is a recently emerged clone of B. pseudomallei and has been shown to be genetically monomorphic with typing methods such as multilocus sequence typing (11). To compare the genetic diversity of our Punjab isolates to that of sequenced strains, we performed a phylogenetic analysis on the 23 loci using the neighbor-joining algorithm in PAUP* 4.0b (9). To determine the genetic relationships among the Punjab population itself, we performed the same analysis using only the Punjab isolates and polymorphic loci (n = 15 loci).
Figure 1
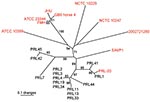
Figure 1. Unrooted neighbor-joining tree based on 23 variable number tandem repeat loci demonstrating that the Punjab isolates (black text and PRL-20) are genetically distinct from and less diverse than available whole genome...
Figure 2
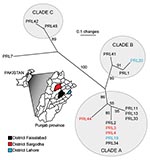
Figure 2. Unrooted neighbor-joining tree showing phylogenetic relationships among 15 samples of Burkholderia mallei from the Punjab Province, Pakistan. Statistical support for each branch derived from 1,000 bootstrap iterations are shown. Sample names...
Combined analysis of the Punjab isolates and WGS showed that the Punjab isolates are phylogenetically distinct from WGS (Figure 1). This finding was also demonstrated in the values for average pairwise distance (APD), where the APD among Punjab isolates is 2× lower than the APD calculated for either the entire phylogeny or the WGS alone (Figure 1). Therefore, the Punjab isolates represent only a small amount of the genetic diversity demonstrated in this pathogen. Phylogenetic analysis of the Punjab isolates alone placed 14 of the 15 samples into 3 distinct clades with 1 sample standing alone (Figure 2). Most samples (9/15) belong to clade A, whose isolates are both temporally and geographically diverse, suggesting that this lineage is ecologically established in Punjab.
Because of the limited sample size, many of the patterns observed from these data may result from sampling bias. However, even a limited amount of genotypic data can be useful in formulating hypotheses regarding the dispersal of B. mallei. For example, the presence of samples from Faisalabad in each clade suggests that this district may be a center of diversity in the province (Figure 2) but this does not indicate a lesser degree of diversity in other districts where fewer samples were collected.
The diversity seen in the district of Faisalabad may result from either 1) the industrial nature of Faisalabad or 2) from high endemism of B. mallei in the region. Currently, the district has ≈10,000 horses and mules and >44,000 donkeys, plus other transient equines (12). Many equines move through and work in the city, potentially introducing strains from surrounding regions. Because horses and mules can be positive but asymptomatic for glanders (13), many hosts are available to maintain strains throughout the region. Communal stables and water troughs are common throughout the district and B. mallei has been isolated from these water troughs (A. Naureen, unpub. data). Furthermore, B. mallei can remain viable in contaminated stables for up to 6 weeks (14) and in sterile tap water for up to 4 weeks (15), which provides an environment for establishment and retention of B. mallei populations in Faisalabad.
Combining phylogenetic with epidemiologic data reveals how B. mallei disseminates throughout a region. For example, epidemiologic data suggests that 2 horses from a farm in the district of Sargodha (PRL3 and PRL4) contracted glanders while at a polo club in the Lahore district. This is supported by VNTR data, as these 2 isolates clustered phylogenetically with one of the samples obtained from an outbreak that occurred at the same polo club 3 months prior (groups 5 and 6, Table). Furthermore, at the time of the PRL3 and PRL4 infections, a co-resident mule with no previous travel history (PRL44) was negative for glanders, making it unlikely that these horses acquired glanders from their farm. This mule was positive for glanders ≈1.5 years later, and the isolate obtained from its infection clustered phylogenetically with the samples from the polo club and Sargodha horses. Therefore, we hypothesize that the infected horses either directly transferred the disease to the mule or they contaminated a source on the farm which subsequently led to the mules infection. Environmental sampling would be required to identify the original infection source for the horses and subsequent transmission route to the mule. Nevertheless, this case shows a strain that was transferred a distance of 168 km, demonstrating that human-mediated movement of equines can influence the distribution of B. mallei genotypes. This case also suggests that a strain can persist for ≈1.5 years.
Other cases in the province demonstrate that infections either stem from similar strains or are caused by multiple strains. For example, samples that were placed in the same epidemiologic group cluster together phylogenetically (groups 1, 2, and 4; Table), indicating communal infections similar to the cases described above. In contrast, epidemiologic group 5 (PRL19 and PRL20) was separated into 2 distinct clades (Figure 2), indicating that this outbreak was caused by multiple strains. Therefore, it should not be assumed that an outbreak of glanders is always caused by a single strain.
Our study suggests that numerous lineages of Burkholderia mallei are present in Punjab, Pakistan, and that these lineages persist across geographic space and time. Despite this, these isolates appear to be genetically distinct from other studied strains. The economics and use of equines likely contribute to the persistence of glanders in this region because modern methods for control of this disease (monitoring and euthanasia) are not viable options. Therefore, other solutions to curbing the spread of glanders need to be identified. We suggest that a focus on finding methods to improve the sanitary conditions of communal water troughs and stables may lead to a practical solution for disease reduction and containment. Finally, our study demonstrates the utility of VNTRs paired with extensive epidemiologic data for analyzing the distribution of B. mallei genotypes throughout endemic regions.
Ms Hornstra is a research project coordinator at the Microbial Genetics and Genomics Center at Northern Arizona University in Flagstaff. Her research interests focus on genetic diversity and evolutionary history of Burkholderia mallei, B. pseudomallei, and Coxiella burnetii.
Acknowledgments
We thank Raymond Auerbach for computational expertise as well as for designing and writing the scripts used for the in silico typing of whole genome sequences.
This study was supported by NIH-NIAID grants U54AI-56359 and U01AI-075568 and by the US Department of Homeland Security S&T CB Division of Bioforensics R&D Program to P.K.
References
- M’Fadyean J. Glanders. J Comp Pathol Ther. 1904;17:295–317.
- Waag DM, DeShazer D. Glanders: new insights into an old disease. In: Lindler LE, Lebeda FJ, Korch GW, editors. Biological weapons defense: infectious diseases and counterbioterrorism. 1st ed. Totowa (NJ): Humana Press Inc.; 2004. p. 209–37.
- Gilad J, Harary I, Dushnitsky T, Schwartz D, Amsalem Y. Burkholderia mallei and Burkholderia pseudomallei as bioterrorism agents: national aspects of emergency preparedness. Isr Med Assoc J. 2007;9:499–503.PubMedGoogle Scholar
- Naureen A, Saqib M, Muhammad G, Hussain MH, Asi MN. Comparative evaluation of Rose Bengal plate agglutination test, mallein test, and some conventional serological tests for diagnosis of equine glanders. J Vet Diagn Invest. 2007;19:362–7.PubMedGoogle Scholar
- Michelle Wong Su Y, Lisanti O, Thibault F, Toh Su S, Loh Gek K, Hilaire V, . Validation of ten new polymorphic tandem repeat loci and application to the MLVA typing of Burkholderia pseudomallei isolates collected in Singapore from 1988 to 2004. J Microbiol Methods. 2009 Mar 25.
- Moore DD. Preparation and analysis of DNA. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, et al., editors. Current protocols in molecular biology. New York: Wiley & Sons; 1995. p. 2.4.1–2.4.2.
- U’Ren JM, Schupp JM, Pearson T, Hornstra H, Friedman CL, Smith KL, et al. Tandem repeat regions within the Burkholderia pseudomallei genome and their application for high resolution genotyping. BMC Microbiol. 2007;7:23. DOIPubMedGoogle Scholar
- NCBI. ENTREZ Genome Project. Burkholderia mallei [cited 2009 Jul 21]. Available from http://www.ncbi.nlm.nih.gov/sites/entrez?db=genomeprj&cmd=search&term=txid13373
- Pearson T, U’Ren JM, Schupp JM, Allan GJ, Foster PG, Mayo MJ, et al. VNTR analysis of selected outbreaks of Burkholderia pseudomallei in Australia. Infect Genet Evol. 2007;7:416–23. DOIPubMedGoogle Scholar
- Girard JM, Wagner DM, Vogler AJ, Keys C, Allender CJ, Drickamer LC, Differential plague-transmission dynamics determine Yersinia pestis population genetic structure on local, regional, and global scales. Proc Natl Acad Sci U S A. 2004;101:8408–13. DOIPubMedGoogle Scholar
- Godoy D, Randle G, Simpson AJ, Aanensen DM, Pitt TL, Kinoshita R, Multilocus sequence typing and evolutionary relationships among the causative agents of melioidosis and glanders, Burkholderia pseudomallei and Burkholderia mallei. J Clin Microbiol. 2003;41:2068–79. DOIPubMedGoogle Scholar
- Pakistan Agriculture Census Organization, Government of Pakistan, Statistics Division. Pakistan livestock census. 2006 [cited 2009 Apr 15]. Available from http://www.statpak.gov.pk/depts/aco/publications/pakistan-livestock-cencus2006/lsc2006.html
- Al-Ani FK, Al-Delaimi AK, Ali AH. Glanders in horses: clinical and epidemiological studies in Iraq. Pakistan Vet J. 1987;7:126–9.
- Henning MW. Glanders, farcy, droes, malleus. Animal diseases in South Africa being an account of the infectious diseases of domestic animals. Third edition (completely revised) including a description of a number of diseases not reported in the previous edition. Johannesburg (South Africa): Central News Agency Ltd.; 1956.
- Miller WR, Pannell L, Cravitz L, Tanner WA, Ingalls MS. Studies on certain biological characteristics of Malleomyces mallei and Malleomyces pseudomallei: I. morphology, cultivation, viability, and isolation from contaminated specimens. J Bacteriol. 1948;55:115–26.
Figures
Table
Cite This ArticleTable of Contents – Volume 15, Number 12—December 2009
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Paul Keim, Northern Arizona University, 1298 S Knoles Drive, Bldg 56, PO Box 4073, Flagstaff, AZ 86011-4073, USA
Top