Volume 15, Number 12—December 2009
Dispatch
Mopeia Virus–related Arenavirus in Natal Multimammate Mice, Morogoro, Tanzania
Figure 1
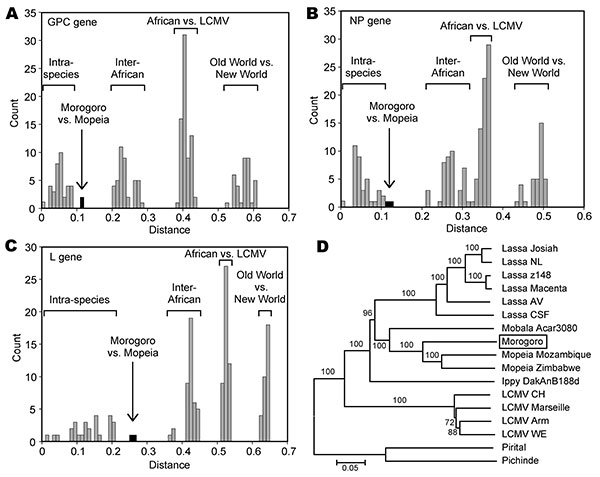
Figure 1. Genetic distances and phylogenetic relationship among arenaviruses, including Morogoro virus. Amino acid sequence diversity was calculated using p distance. Full-length glycoprotein precursor (GPC), nucleoprotein (NP), and large (L) gene amino acid sequences of the following arenaviruses were pairwise compared: Lassa virus (strains Josiah, NL, Z148, Macenta, AV, and CSF), Mobala Acar3080, Morogoro 3017/2004, Mopeia virus (strains Mozambique and Zimbabwe), Ippy DakAnB188d, lymphocytic choriomeningitis virus (LCMV) (strains CH-5692, Marseille, Armstrong, and WE for all genes; Traub and Pasteur for GPC and NP only), Pirital, and Pichinde. Frequency histograms of pairwise distances are shown for A) GPC gene; B) NP gene; and C) L gene. The ranges for intraspecies distances (i.e., pairwise differences between strains of the same virus species); distances between different African arenavirus species; between African arenaviruses and LCMV; and between Old World and New World viruses are marked above the bars. Bars representing the distances between Morogoro virus and the most closely related viruses (Mopeia virus strains) are filled in black. D) Phylogeny of Old World arenaviruses based on full-length L gene amino acid sequences. The tree was inferred by using the neighbor-joining method implemented in the MEGA software package (www.megasoftware.net). The New World arenaviruses Pirital and Pichinde were used as outgroups. Numbers represent bootstrap support (1,000 replications). Identical trees with respect to the phylogenetic position of Morogoro virus (shown in the box) were obtained with full-length GPC and NP amino acid sequences (not shown). Scale bar indicates nucleotide substitutions per site.