Volume 15, Number 12—December 2009
Dispatch
Dobrava-Belgrade Virus Spillover Infections, Germany
Figure 2
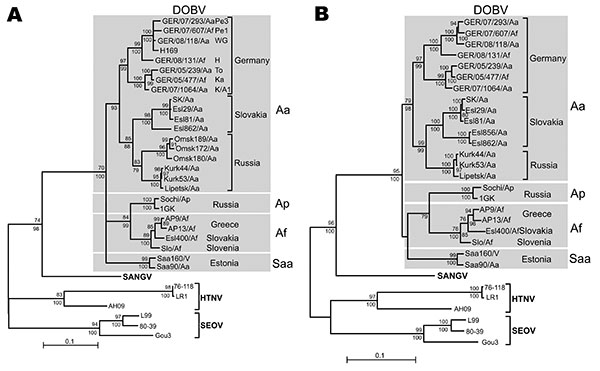
Figure 2. Maximum-likelihood (ML) phylogenetic trees of Dobrava-Belgrade virus (DOBV) based on partial small (S) segment nucleotide sequences of 559 nt (position 377–935) (A) and complete nucleocapsid protein coding nucleotide sequences (S segment open reading frame) (B). The ML trees (Tamura-Nei evolutionary model) were calculated using TREE-PUZZLE package (www.tree-puzzle.de). Scale bars indicate an evolutionary distance of 0.1 substitutions per position in the sequence. Values above the branches represent PUZZLE support values. Values below the branches are bootstrap values of the corresponding neighbor-joining tree (Tamura-Nei evolutionary model) calculated with the PAUP* software (paup.csit.fsu.edu) from 10,000 bootstrap pseudoreplicates. Only values >70% (considered significant) are shown. Different DOBV lineages are indicated by gray boxes. SANGV, Sangassou virus; HTNV, Hantaan virus; SEOV, Seoul virus; Saa, Saaremaa virus; Aa, Apodemus agrarius; Ap, A. ponticus; Af, A. flavicollis. WG, district Lüneburg, Lower Saxony (LS); Pe1 and Pe3, district Güstrow; H, district Nordvorpommern, K/A1, district Demmin, all Mecklenburg-Western Pomerania (MWP); To and Ka, district Ostprignitz-Ruppin, Brandenburg (BB). Before tree construction, automated screening for recombination between the S segment sequences was performed using program RDP3 (15), which used 6 recombination detection programs: Bootscan, Chimeric, GENECONV, MaxChi, RDP, and SiScan with their default parameters. No putative recombinant regions could be conclusively detected by >3 programs and subsequently verified by phylogenetic trees.
References
- Avsic-Zupanc T, Petrovec M, Furlan P, Kaps R, Elgh F, Lundkvist A. Hemorrhagic fever with renal syndrome in the Dolenjska region of Slovenia–a 10-year survey. Clin Infect Dis. 1999;28:860–5. DOIPubMedGoogle Scholar
- Krüger DH, Ulrich R, Lundkvist Å. Hantavirus infections and their prevention. Microbes Infect. 2001;3:1129–44. DOIPubMedGoogle Scholar
- Klempa B, Tkachenko EA, Dzagurova TK, Yunicheva YV, Morozov VG, Okulova NM, Hemorrhagic fever with renal syndrome caused by 2 lineages of Dobrava hantavirus, Russia. Emerg Infect Dis. 2008;14:617–25. DOIPubMedGoogle Scholar
- Sibold C, Ulrich R, Labuda M, Lundkvist Å, Martens H, Schütt M, Dobrava hantavirus causes hemorrhagic fever with renal syndrome in central Europe and is carried by two different Apodemus mice species. J Med Virol. 2001;63:158–67. DOIPubMedGoogle Scholar
- Klempa B, Stanko M, Labuda M, Ulrich R, Meisel H, Krüger DH. Central European Dobrava hantavirus isolate from striped field mouse, Apodemus agrarius. J Clin Microbiol. 2005;43:2756–63. DOIPubMedGoogle Scholar
- Sjölander KB, Golovljova I, Vasilenko V, Plyusnin A, Lundkvist A. Serological divergence of Dobrava and Saaremaa hantaviruses: evidence for two distinct serotypes. Epidemiol Infect. 2002;128:99–103.PubMedGoogle Scholar
- Meisel H, Lundkvist Å, Gantzer K, Bär W, Sibold C, Krüger DH. First case of infection with hantavirus Dobrava in Germany. Eur J Clin Microbiol Infect Dis. 1998;17:884–5. DOIPubMedGoogle Scholar
- Klempa B, Schütt M, Auste B, Ulrich R, Meisel H, Krüger DH. First molecular identification of human Dobrava virus infection in Central Europe. J Clin Microbiol. 2004;42:1322–5. DOIPubMedGoogle Scholar
- Essbauer S, Schmidt J, Conraths FJ, Friedrich R, Koch J, Hautmann W, A new Puumala hantavirus subtype in rodents associated with an outbreak of nephropathia epidemica in South-East Germany in 2004. Epidemiol Infect. 2006;134:1333–44. DOIPubMedGoogle Scholar
- Kocher TD, Thomas WK, Meyer A, Edwards SV, Pääbo S, Villablan CA, Dynamics of mitochondrial DNA evolution in animals: Amplification and sequencing with conserved primers. Proc Natl Acad Sci U S A. 1989;86:6196–200. DOIPubMedGoogle Scholar
- Mitchell-Jones AJ, Amori G, Bogdanowicz W, Krystufek B, Reijnders PJH, Spitzenberger F, The atlas of European mammals. London: Academic Press; 1999.
- Klingström J, Heyman P, Escutenaire S, Sjölander KB, De Jaegere F, Henttonen H, Rodent host specificity of European hantaviruses: evidence of Puumala virus interspecific spillover. J Med Virol. 2002;68:581–8. DOIPubMedGoogle Scholar
- Avsic-Zupanc T, Nemirov K, Petrovec M, Trilar T, Poljak M, Vaheri A, Genetic analysis of wild-type Dobrava hantavirus in Slovenia: co-existence of two distinct genetic lineages within the same natural focus. J Gen Virol. 2000;81:1747–55.PubMedGoogle Scholar
- Weidmann M, Schmidt P, Vackova M, Krivanec K, Munclinger P, Hufert FT. Identification of genetic evidence for Dobrava virus spillover in rodents by nested reverse transcription (RT)-PCR and TaqMan RT-PCR. J Clin Microbiol. 2005;43:808–12. DOIPubMedGoogle Scholar
- Martin DP, Williamson C, Posada D. RDP2: recombination detection and analysis from sequence alignments. Bioinformatics. 2005;21:260–2. DOIPubMedGoogle Scholar