Volume 15, Number 2—February 2009
Research
Epidemiology of Vibrio parahaemolyticus Outbreaks, Southern Chile
Cite This Article
Citation for Media
Abstract
Disease outbreaks caused by Vibrio parahaemolyticus in Puerto Montt, Chile, began in 2004 and reached a peak in 2005 at 3,600 clinical cases. Until 2006, every analyzed case was caused by the serovar O3:K6 pandemic strain. In the summer of 2007, only 475 cases were reported; 73% corresponded to the pandemic strain. This decrease was associated with a change in serotype of many pandemic isolates to O3:K59 and the emergence of new clinical strains. One of these strains, associated with 11% of the cases, was genotypically different from the pandemic strain but contained genes that were identical to those found on its pathogenicity island. These findings suggest that pathogenicity-related genes were laterally transferred from the pandemic strain to one of the different V. parahaemolyticus groups comprising the diverse and shifting bacterial population in shellfish in this region.
In 1998 in Antofagasta, Chile (23°39′S, 70°24′W), ≈300 human cases of infection with Vibrio parahaemolyticus caused by consumption of contaminated seafood were reported (1). Outbreaks have not been observed in this region since 1998. During 2004–2007, ≈7,000 cases were reported farther south in Puerto Montt (41°29′S, 72°24′W) (2–5). However, outbreaks generally have been decreasing; there were ≈1,500 cases in 2004, 3,600 in 2005, 900 in 2006, and 475 in 2007 (6) (http://epi.minsal.cl/epi/html/Actualidad/Vibrio.htm).
Until 2006, ≈100% of the cases analyzed were caused by a clonal group originally observed in Southeast Asia in 1996 (4,5,7). This group was known as the V. parahaemolyticus pandemic strain because it had reached coastal environments worldwide and caused outbreaks. It belongs to the O3:K6 serovar, although at least 21 serovariants have emerged since 1996 (8). These serovariants also have specific sequences corresponding to genes such as toxRS/new (9), open reading frame 8 (orf8) (10), and tdh, but lack others such as trh, which is found in other pathogenic strains.
Genome sequencing of the RIMD2210633 pandemic strain showed that it has 2 sets of gene clusters that encode the type III secretion system (TTSS) apparatus (11). This apparatus is used by several gram-negative pathogenic bacteria to secrete and translocate virulence factor proteins into the cytosol of eukaryotic cells (12). TTSS1 is involved in cytotoxicity against HeLa cells and TTSS2 is involved in enterotoxic activity in a rabbit ileal loop test (13). The first cluster is located on the large chromosome, and the second is located on the small chromosome. The second cluster contains 2 copies of the tdh gene and is located on a pathogenicity island (a discrete genetic unit that contains genes responsible for pathogenicity and virulence) probably obtained by recent lateral transfer (11). TTSS2 has been found only in strains showing β-type hemolysis on a specialized blood agar medium known as Wagatsuma agar (11). This hemolysis is called the Kanagawa phenomenon and is considered a useful marker for identification of pathogenic strains. Recently, TTSS genes related to the TTSS2 cluster were reported in clinical and environmental non-O1, non-O139 V. cholerae strains (14).
The clonal nature of the pandemic V. parahaemolyticus isolates was ascertained by the similarity of patterns obtained by genome restriction fragment length polymorphism–pulsed-field gel electrophoresis (15), arbitrarily primed PCR (7,9), direct genome restriction enzyme analysis (DGREA) (4), and multilocus sequence typing (MLST) (16,17). However, the pandemic strain is a minor fraction of a diverse and shifting V. parahaemolyticus population found in shellfish in Puerto Montt (2). In an effort to understand the epidemiology of these outbreaks, we studied V. parahaemolyticus isolates obtained from human cases and shellfish during the summer of 2007. Our results indicate replacement of the O3:K6 pandemic strain by new serotype O3:K59 and new pathogenic V. parahaemolyticus groups. The decrease in the number of clinical cases may have been caused by diminution of the V. parahaemolyticus O3:K6 pandemic group in regional seafood.
Strains
V. parahaemolyticus RIMD 2210633 (VpKX) and RIMD 2210086 (VpI) were obtained from the Research Institute for Microbial Diseases, Osaka University, Osaka, Japan. The Chilean environmental strains, identified as PMA with a number according to origin and year of isolation, were obtained from shellfish samples obtained during outbreaks from 2004 though 2007. Most strains have been described (4). PMC38.7, PMC60.7, PMC53.7, and PMC75.7 are isolates from clinical samples obtained in 2007. Each of these isolates corresponds to the type isolate of the 23 groups differentiated by DGREA as described (4) and reported in this article.
Analysis
Samples of clinical cases and shellfish were obtained and analyzed as described (4). Isolation, growth, and characterization of isolates, including their DGREA patterns, were conducted as described (4,5). Each DGREA pattern found in 2007 was compared with those for previous years. When similarities in patterns were observed, their identity was checked by comparing patterns obtained in the same electrophoretic analysis.
PCR assays were performed by using ≈10 ng of total bacterial DNA per reaction tube. Amplification conditions were those previously reported for tlh, tdh, and trh (18), orf8 (19), and toxRS/new (9) genes. T3SS2 genes (VPA1335, VPA1338, VPA1339, VPA1341, VPA1342, VPA1346, VPA1349, VPA1354, VPA1355, VPA1362, and VPA1367) were amplified by using primers reported for a microarray assay at 61ºC by Meador et al (20). Genes VPA1321 and VPA1376, which are located at the extremes of the pathogenicity island, were amplified by using primers designed using the Primer3 program (http://primer3.sourceforge.net). Sequences of these primers were VPA1321f: 5′-TGACATGCACGGCAATAGAT-3′, VPA1321r: 5-ACAGAGTTGGTTTCGCAGGT-3′, VPA1376 f: 5′-CATCGAGCGATCTTTCACAA-3′, and VPA1376r: 5′-ACCGGTTTCCAACCTTCTCT-3′. Housekeeping genes for MLST were amplified by using primers for V. parahaemolyticus (17) and from the MLST website (http://pubmlst.org/vparahaemolyticus) developed by Keith Jolley (University of Oxford, Oxford, UK) (21).
PCR products were purified by using either the Wizard SV Gel or PCR Clean-Up Systems (Promega, Madison, WI, USA) and sequenced in both directions by Macrogen (Seoul, South Korea) or by McLAB (South San Francisco, CA, USA) by using forward and reverse amplification primers or primers M13F and M13R (MLST loci). DNA sequences were analyzed individually and manually assembled. Alignments and sequence similarities were obtained by using BioEdit software (22). Sequences obtained were deposited in GenBank under accession nos. EU185060–EU185092.
Amplification and sequencing of the variable region of the 16S rRNA (rrs) between nucleotides 357 and 518 (Escherichia coli numbering) were performed as described (23). This analysis consisted of separation of rrs alleles in PMC38.7 by pulsed-field gel electrophoresis and PCR amplification of the variable region in excised bands as described (24).
V. parahaemolyticus Associated with Human Cases in 2007
Figure 1
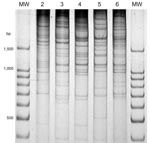
Figure 1. Direct genome restriction enzyme analysis with NaeI of clinical isolates of Vibrio parahaemolyticus representative of the 5 patterns observed during the outbreaks in Puerto Montt, Chile, January and February, 2007. Lanes...
V. parahaemolyticus isolates from 37 human case-patients with diarrhea from the summer of 2007 in the Puerto Montt region were analyzed and grouped according to serotype, presence of genetic markers (orf8, toxRS/new, tlh, tdh, trh) and distinctiveness of their DGREA patterns (Table 1, Figure 1). One isolate from each patient was characterized. On the basis of genetic markers and DGREA pattern, isolates from 27 patients corresponded to the pandemic clonal group. However, 40% of the 20 serotyped strains of this group contained a K59 capsular antigen instead of the characteristic K6 antigen, and 25% cross-reacted with antisera for K6 and K59 antigens. Another difference from strains of previous years was that the relative number of cases associated with the pandemic strain (73%) was lower than the 100% observed in previous summers (4). Isolates from the other 27% of patients with clinical cases lacked the characteristic markers of the pandemic strains, i.e., orf8 and toxRS/new, and had 4 new DGREA groups. One of these groups contained 4 tdh-positive isolates (11% of cases). A second group contained 4 isolates positive for tdh and trh genes. The other 2 groups contained 1 isolate each, and both were negative for these 2 markers (Table 1). One isolate chosen as the type strain of each group was typed by MLST (17). MLST sequence type corresponded with groups determined by other analyzed genetic properties (Table 1).
Characterization of Nonpandemic Strains
Strains positive for tdh, other than the pandemic strain, had not been isolated from patients with clinical cases in Puerto Montt during the 3 previous summer outbreaks. It has been reported that this gene may be spread by insertion sequence–like elements (25,26). The possibility that tdh found in nonpandemic strains was derived from the pandemic strain was explored. PCR amplicons of the tdh gene of the 2 nonpandemic groups was sequenced in isolates designated as type strains for each group: PMC60.7 for the group containing tdh and trh and PMC38.7 for the group containing only tdh. The amplicon of isolate PMC60.7 had an identical sequence to that reported for tdhA except for 1 nucleotide (11). Conversely, PMC38.7 had an identical sequence to that expected for a mixture of the 2 tdh genes in VpKX. These 2 genes differ slightly and the mixture of their PCR products should show polymorphisms in specific sites (11).
This observation suggested the presence of tdhS and tdhA genes in PMC38.7 with identical sequences to those found in the pandemic strain. Because these 2 genes are located close to each end of the pathogenicity island in chromosome 2 of the pandemic V. parahaemolyticus (11), the presence of the entire island was explored by PCR amplification of 11 genes of TTSS2 located in the island and of genes VPA1321 and VPA1376 located at the extremes of the island near tdhA and tdhS, respectively. Each tested gene was found in the PMC38.7 strain, and sequences of their PCR products were identical to those reported for the pandemic strain genes, except for 1 nucleotide in VPA1342. Serotyping of PMC38.7 indicated an O10:K20 serovar. Because MC38.7 is genetically different from the pandemic strain (Table 1, Figure 1), the high degree of homology of these genes suggested that the entire pathogenicity island had recently been transferred from the pandemic strain.
PMC38.7 also differs from the pandemic strain and most clinical isolates by the presence of intragenomic heterogeneity among its multiple 16S rRNA genes, a feature seldom observed in clinical isolates but frequently observed among environmental isolates. This finding is probably caused by lateral transfer of rrs (23). Three rrs genes, with sequences corresponding to V. parahaemolyticus groups VpD1-B4, ATA65-B2, and VpKX-AB (23), were observed in PMC38.7.
V. parahaemolyticus Associated with Shellfish
Figure 2
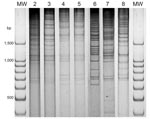
Figure 2. Direct genome restriction enzyme analysis with NaeI of Vibrio parahaemolyticus isolates from shellfish collected in Puerto Montt, Chile, summer, 2007. Gel shows representative strains for every observed pattern. Patterns of groups...
Figure 3

Figure 3. Number of seafood samples containing Vibrio parahaemolyticus corresponding to different direct genome restriction enzyme analysis (DGREA) groups observed in Puerto Montt, Chile, each summer, 2004–2007.
There are a large number of V. parahaemolyticus strains in the environment in the Puerto Montt region. Only the pandemic strain was isolated from clinical samples before and during the summer of 2006, but 20 different strains were isolated from shellfish during that period (2). Characterization of 52 isolates from 20 shellfish samples during the summer of 2007 indicated ≥5 DGREA groups; 3 of them were not previously observed (Table 2, Figure 2). This finding increases the number of different strains found in shellfish of the region to 23. Figure 3 shows the number of shellfish samples containing each of the 23 DGREA groups found during the past 4 years, including the summer of 2007 (2 and this study). Only 4 of the 20 shellfish samples examined produced tdh-positive enrichment cultures, which is indicative of pathogenic strains. However, no tdh-positive isolates were obtained from these enrichments after plating on thiosulfate citrate bile salts sucrose agar.
T3SS2 Genes in Other Nonpandemic V. parahaemolyticus Strains
Because the presence of TTSS2 genes is not exclusive to the pandemic strain (20), their occurrence in the other environmental and clinical V. parahaemolyticus DGREA groups found in Puerto Montt was explored. We analyzed by PCR amplification 20 strains corresponding to 18 environmental DGREA groups (PMA79, 112, 118, 189, 337, 339, 3316, 1.5, 19.5, 22.5, 27.5, 13.6, 34.6, 36.6, 40.6, 1.7, 11.7, and 21.7) and 1 strain from each of 3 new clinical groups (PMC60.7, 53.7, and 75.7) found in Puerto Montt in 2007 for the VPA1335 gene (found in T3SS2). Among these strains, only PMA339, isolated from shellfish in the summer of 2004, was positive. PCR amplification of PMA339 for the other genes in the pathogenicity island showed positive reactions for all the genes tested in PMC38.7. Nevertheless, sequences of amplicons showed strong differences from those found in PMC38.7 and those reported for the pandemic strain genome. Similarity ranged from 99.4% for VPA1362 to 93.8% for VPA1346; the average for all genes tested was 97.7%. These differences indicate a much larger evolutionary distance between the T3SS2 genes in PMA339 and the pandemic strain than between PMC38.7 and the pandemic strain.
The epidemiology of outbreaks caused by V. parahaemolyticus in the Puerto Montt region is changing. The number of clinical cases caused by the pandemic strain has decreased, accompanied by a change of serotype from O3:K6 to O3:K59. The changing serotype of the pandemic strain has been recently reviewed by Nair et al (8). These authors showed that the more recent serotypes do not have the propensity for increasing hospital admissions observed with O3:K6, and some type of change in the epidemic process seems to be evident. Genes for the biosynthesis of capsular polysaccharides, which are major antigens (K) of V. parahaemolyticus, are probably encoded in a gene cluster characterized by variability that may occur through lateral gene transfer (27). Isolates reacting with antisera for K6 and K59 antigens may have changed part of the K epitopes reacting with the commercial polyclonal antiserum.
The percentage of clinical cases caused by the pandemic strain decreased from 100% in 2006 to 73% in 2007. Four clinical strains, not previously observed, emerged in 2007. Among these, 1 group representing 11% of the clinical cases, with type strain PMC38.7, may have recently received the genes on the pathogenicity island of the pandemic strains. Pathogenicity island genes identical to those in the pandemic strain in this bacterial group and differences in housekeeping genes and DGREA patterns are best explained by transfer of the pathogenicity island from the pandemic strain to an indigenous strain. The indigenous V. parahaemolyticus population in shellfish is diverse, and the predominant strains seem to change every year. A detailed examination of the putative genomic island in PMC38.7, its integration site, and its flanking regions, will probably help differentiate among possible mechanisms of DNA transfer.
The presence of TTSS2 genes is not exclusive of the pandemic and PMC38.7 strain; they were also found in an environmental isolate of V. parahaemolyticus (PMA339). However, PMA339 has not been observed among clinical isolates. TTSS2 genes have been found in other V. parahaemolyticus clinical strains (20); however, we identified them in environmental strains. Although on the basis of sequences obtained from their amplicons, TTSS2 genes in PMA339 seem to have independently evolved from the pandemic strain over a considerable time, they are still more closely related to the TTSS2 of V. parahaemolyticus than to those recently found in non-O1 or non-O139 V. cholerae (14).
Little is known of the origin of the other 3 clinical groups (60.7, 1.5, and 75.7). Group 60.7 contains tdh and trh, but this tdh does not seem to be derived from the pandemic strain. Strains of groups 1.5 and 75.7 lack both pathogenicity-associated genes, but finding these isolates in patients is not unusual (28). PMC75.7, a clinical strain, contains a recA gene that is closely related to that of PMA339, the environmental isolate containing TTSS2 genes (17 and http://pubmlst.org/vparahaemolyticus). However, in view of a recent report (29), one should consider that the 2 human isolates lacking tdh or trh genes may correspond to nonvirulent strains that proliferate during infection with a virulent strain.
The abundance and frequency of pandemic and nonpandemic V. parahaemolyticus in shellfish seem to have been less in 2007 than in previous years. In 2006, 10 of 20 shellfish samples were positive for tdh after enrichment; in 2007, only 4 of 20 samples were positive. PCR amplification of colonies obtained after plating the enrichment culture on thiosulfate citrate bile salts sucrose agar enabled identification of tdh-positive colonies in 6 samples in 2006; none could be identified by the same method in 2007. The observed decrease in outbreaks was probably caused by a decrease in raw seafood consumption as a result of a public health campaign and a decrease in the load of the highly virulent pandemic strain in shellfish. However, this tendency could change on the basis of dispersion and virulence of emerging pathogenic strains.
Dr Harth is a postdoctoral fellow in the Department of Vaccinology and Applied Microbiology at the Helmholtz Centre of Infection Research, Braunschweig, Germany. Her research interests include the epidemiology and evolution of pathogenic bacteria.
Acknowledgments
We thank Katherine García, Paulina Uribe, Gastón Higuera, and Beatriz Zabala for help with sample collection and isolation and characterization of bacterial strains.
This study was supported in part by grants from Fondo Nacional de Desarrollo Científico Technológico (nos. 1040875 and 1070658). E.H. was supported by a scholarship from Deutscher Akademischer Austausch Diemst.
References
- Cordova JL, Astorga J, Silva W, Riquelme C. Characterization by PCR of Vibrio parahaemolyticus isolates collected during the 1997–1998 Chilean outbreak. Biol Res. 2002;35:433–40.PubMedGoogle Scholar
- Fuenzalida L, Armijo L, Zabala B, Hernandez C, Rioseco ML, Riquelme C, Vibrio parahaemolyticus strains isolated during investigation of the summer 2006 seafood related diarrhea outbreaks in two regions of Chile. Int J Food Microbiol. 2007;117:270–5. DOIPubMedGoogle Scholar
- Cabello FC, Espejo RT, Hernandez MC, Rioseco ML, Ulloa J, Vergara JA. Vibrio parahaemolyticus O3:K6 epidemic diarrhea, Chile, 2005. Emerg Infect Dis. 2007;13:655–6.PubMedGoogle Scholar
- Fuenzalida L, Hernandez C, Toro J, Rioseco ML, Romero J, Espejo RT. Vibrio parahaemolyticus in shellfish and clinical samples during two large epidemics of diarrhoea in southern Chile. Environ Microbiol. 2006;8:675–83. DOIPubMedGoogle Scholar
- Gonzalez-Escalona N, Cachicas V, Acevedo C, Rioseco ML, Vergara JA, Cabello F, Vibrio parahaemolyticus diarrhea, Chile, 1998 and 2004. Emerg Infect Dis. 2005;11:129–31.PubMedGoogle Scholar
- Olea AM, González C, Chiu M, Vallebuona C, Labraña M, Martiniello F. Brote de gastroenteritis por Vibrio parahaemolyticus en Chile. Revista Chilena Salud Pública. 2005;9:51–3.
- Okuda J, Ishibashi M, Hayakawa E, Nishino T, Takeda Y, Mukhopadhyay AK, Emergence of a unique O3:K6 clone of Vibrio parahaemolyticus in Calcutta, India, and isolation of strains from the same clonal group from Southeast Asian travelers arriving in Japan. J Clin Microbiol. 1997;35:3150–5.PubMedGoogle Scholar
- Nair GB, Ramamurthy T, Bhattacharya SK, Dutta B, Takeda Y, Sack DA. Global dissemination of Vibrio parahaemolyticus serotype O3:K6 and its serovariants. Clin Microbiol Rev. 2007;20:39–48. DOIPubMedGoogle Scholar
- Matsumoto C, Okuda J, Ishibashi M, Iwanaga M, Garg P, Rammamurthy T, Pandemic spread of an O3:K6 clone of Vibrio parahaemolyticus and emergence of related strains evidenced by arbitrarily primed PCR and toxRS sequence analyses. J Clin Microbiol. 2000;38:578–85.PubMedGoogle Scholar
- Nasu H, Iida T, Sugahara T, Yamaichi Y, Park KS, Yokoyama K, A filamentous phage associated with recent pandemic Vibrio parahaemolyticus O3:K6 strains. J Clin Microbiol. 2000;38:2156–61.PubMedGoogle Scholar
- Makino K, Oshima K, Kurokawa K, Yokoyama K, Uda T, Tagomori K, Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V. cholerae. Lancet. 2003;361:743–9. DOIPubMedGoogle Scholar
- Hueck CJ. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev. 1998;62:379–433.PubMedGoogle Scholar
- Park KS, Ono T, Rokuda M, Jang MH, Okada K, Iida T, Functional characterization of two type III secretion systems of Vibrio parahaemolyticus. Infect Immun. 2004;72:6659–65. DOIPubMedGoogle Scholar
- Dziejman M, Serruto D, Tam VC, Sturtevant D, Diraphat P, Faruque SM, Genomic characterization of non-O1, non-O139 Vibrio cholerae reveals genes for a type III secretion system. Proc Natl Acad Sci U S A. 2005;102:3465–70. DOIPubMedGoogle Scholar
- Wong HC, Liu SH, Wang TK, Lee CL, Chiou CS, Liu DP, Characteristics of Vibrio parahaemolyticus O3:K6 from Asia. Appl Environ Microbiol. 2000;66:3981–6. DOIPubMedGoogle Scholar
- Chowdhury NR, Stine OC, Morris JG, Nair GB. Assessment of evolution of pandemic Vibrio parahaemolyticus by multilocus sequence typing. J Clin Microbiol. 2004;42:1280–2. DOIPubMedGoogle Scholar
- González-Escalona N, Martinez-Urtaza J, Romero J, Espejo RT, Jaykus LA, DePaola A. Determination of molecular phylogenetics of Vibrio parahaemolyticus strains by multilocus sequence typing. J Bacteriol. 2008;190:2831–40. DOIPubMedGoogle Scholar
- Bej AK, Patterson DP, Brasher CW, Vickery MC, Jones DD, Kaysner CA. Detection of total and hemolysin-producing Vibrio parahaemolyticus in shellfish using multiplex PCR amplification of tl, tdh and trh. J Microbiol Methods. 1999;36:215–25. DOIPubMedGoogle Scholar
- Laohaprertthisan V, Chowdury A, Kongmuang U, Kalnauwakul S, Ishibashi M, Matsumoto C, Prevalence and serodiversity of the pandemic clone among the clinical strains of Vibrio parahaemolyticus isolated in southern Thailand. Epidemiol Infect. 2003;130:395–06.PubMedGoogle Scholar
- Meador CE, Parsons MM, Bopp CA, Gerner-Smidt P, Painter JA, Vora GJ. Virulence gene– and pandemic group–specific marker profiling of clinical Vibrio parahaemolyticus isolates. J Clin Microbiol. 2007;45:1133–9. DOIPubMedGoogle Scholar
- Jolley KA, Chan MS, Maiden MC. mlstdbNet-distributed multi-locus sequence typing (MLST) databases. BMC Bioinformatics. 2004;5:86. DOIPubMedGoogle Scholar
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–8.
- Harth E, Romero J, Torres R, Espejo RT. Intragenomic heterogeneity and intergenomic recombination among Vibrio parahaemolyticus 16S rRNA genes. Microbiology. 2007;153:2640–7. DOIPubMedGoogle Scholar
- Gonzalez-Escalona N, Romero J, Espejo RT. Polymorphism and gene conversion of the 16S rRNA genes in the multiple rRNA operons of Vibrio parahaemolyticus. FEMS Microbiol Lett. 2005;246:213–9. DOIPubMedGoogle Scholar
- Terai A, Baba K, Shirai H, Yoshida O, Takeda Y, Nishibuchi M. Evidence for insertion sequence-mediated spread of the thermostable direct hemolysin gene among Vibrio species. J Bacteriol. 1991;173:5036–46.PubMedGoogle Scholar
- Nishibuchi M, Kaper JB. Thermostable direct hemolysin gene of Vibrio parahaemolyticus: a virulence gene acquired by a marine bacterium. Infect Immun. 1995;63:2093–9.PubMedGoogle Scholar
- Izutsu K, Kurokawa K, Tashiro K, Kuhara S, Hayashi T, Honda T, Comparative genomic analysis using microarray demonstrates a strong correlation between the presence of the 80-kilobase pathogenicity island and pathogenicity in Kanagawa phenomenon-positive Vibrio parahaemolyticus strains. Infect Immun. 2008;76:1016–23. DOIPubMedGoogle Scholar
- Honda T, Iida T. The pathogenicity of Vibrio parahaemolyticus and the role of the thermostable direct haemolysin and related haemolysin. Reviews in Medical Microbiology. 1993;4:106–13.
- Bhoopong P, Palittapongarnpim P, Pomwised R, Kiatkittipong A, Kamruzzaman M, Nakaguchi Y, Variability of properties of Vibrio parahaemolyticus strains isolated from individual patients. J Clin Microbiol. 2007;45:1544–50. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 15, Number 2—February 2009
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Romilio T. Espejo, Instituto de Nutrición y Tecnología de los Alimentos, Universidad de Chile, El Líbano 5524, Macul, Santiago 6903625, Chile;
Top