Volume 15, Number 7—July 2009
Dispatch
Genetically Diverse Coronaviruses in Wild Bird Populations of Northern England
Figure
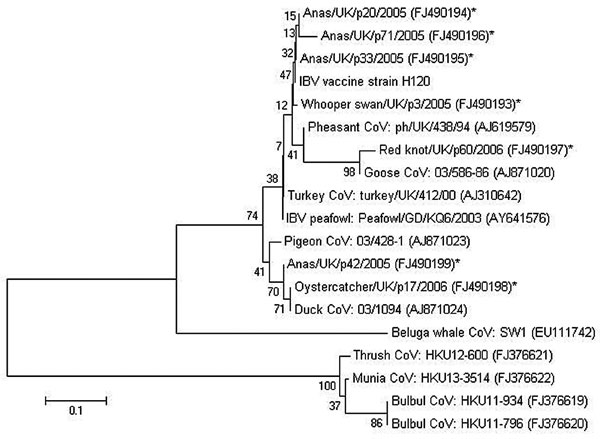
Figure. Minimum-evolution tree (11) of coronaviruses based on a 146-bp fragment of the 3′ untranslated region of infectious bronchitis virus (IBV). Evolutionary distances were computed by using the Tamura-Nei method (12) and are in the units of the number of base substitutions per site. Coronaviruses detected in wild birds by this study are denoted with an asterisk. Previously published coronavirus sequences from different sources were included for comparative purposes. GenBank accession numbers are shown in brackets. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) is shown next to the branches (13). The tree is drawn to scale; branch lengths are in the same units as those of the evolutionary distances used to infer the phylogenetic tree. Phylogenetic analyses were conducted in MEGA4 (10). CoV, coronavirus. Scale bar indicates nucleotide substitutions per site.
References
- Cavanagh D, Gelb J. Infectious bronchitis. In: Saif YM, editor. Diseases of poultry, 12th ed. Ames (IA): Wiley-Blackwell Publishing; 2008. p. 101–20.
- Worthington KJ, Currie RJ, Jones RC. A reverse transcriptase–polymerase chain reaction survey of infectious bronchitis virus genotypes in Western Europe from 2002 to 2006. Avian Pathol. 2008;37:247–57. DOIPubMedGoogle Scholar
- Adzhar A, Gough RE, Haydon D, Shaw K, Britton P, Cavanagh D. Molecular analysis of the 793/B serotype of infectious bronchitis virus in Great Britain. Avian Pathol. 1997;26:625–40. DOIPubMedGoogle Scholar
- Liu S, Chen J, Chen J, Kong X, Shao Y, Han Z, Isolation of avian infectious bronchitis coronavirus from domestic peafowl (Pavo cristatus) and teal (Anas). J Gen Virol. 2005;86:719–25. DOIPubMedGoogle Scholar
- Sun L, Zhang GH, Jiang JW, Fu JD, Ren T, Cao WS, A Massachusetts prototype-like coronavirus isolated from wild peafowls is pathogenic to chickens. Virus Res. 2007;130:121–8. DOIPubMedGoogle Scholar
- Woo PC, Lau SK, Lam CS, Lai KK, Huang Y, Lee P, Comparative analysis of complete genome sequences of three avian coronaviruses reveals a novel group 3c coronavirus. J Virol. 2008;83:908–17. DOIPubMedGoogle Scholar
- Jonassen CM, Kofstad T, Larsen IL, Lovland A, Handeland K, Follestad A, Molecular identification and characterization of novel coronaviruses infecting graylag geese (Anser anser), feral pigeons (Columbia livia) and mallards (Anas platyrhynchos). J Gen Virol. 2005;86:1597–607. DOIPubMedGoogle Scholar
- Redfern CPE, Clark JA. Ringer’s manual. Thetford (UK): British Trust for Ornithology; 2001.
- Cavanagh D, Mawditt K, Sharma M, Drury SE, Ainsworth HL, Britton P, Detection of a coronavirus from turkey poults in Europe genetically related to infectious bronchitis virus of chickens. Avian Pathol. 2001;30:355–68. DOIPubMedGoogle Scholar
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–9. DOIPubMedGoogle Scholar
- Rzhetsky A, Nei M. A simple method for estimating and testing minimum evolution trees. Mol Biol Evol. 1992;9:945–67.
- Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10:512–26.PubMedGoogle Scholar
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–91. DOIGoogle Scholar
- Williams CJ, Moffitt CM. A critique of methods of sampling and reporting pathogens in populations of fish. J Aquat Anim Health. 2001;13:300–9. DOIGoogle Scholar
- Ambali AG, Jones RC. Early pathogenesis in chicks of infection with an enterotropic strain of infectious bronchitis virus. Avian Dis. 1990;34:809–17. DOIPubMedGoogle Scholar