Volume 16, Number 12—December 2010
Dispatch
Multispacer Typing of Bartonella henselae Isolates from Humans and Cats, Japan
Cite This Article
Citation for Media
Abstract
To determine genotypic distribution of and relationship between human and cat strains of Bartonella henselae, we characterized 56 specimens using multispacer typing (MST). Of 13 MST genotypes identified, 12 were grouped into cluster 1. In Japan, human infections can be caused by B. henselae strains in cluster 1.
The causative agent of cat-scratch disease (CSD), Bartonella henselae, is a gram-negative bacterium associated with cats. Human infection usually occurs through scratches or bites by infected cats and typically is seen with localized lymphadenopathy. Occasionally, the infection may have an atypical manifestation, such as endocarditis, encephalopathy, neuroretinitis, or systemic CSD with hepatic and splenic granuloma (1).
B. henselae strains are classified into two 16S rRNA genotypes, 16S type I/Houston-1 and 16S type II/Marseille. Although both genotypes are present worldwide, 16S type II appears to be dominant in the cat population of Europe, whereas 16S type I is more common in Asia, including Japan (2,3).
Multispacer typing (MST) is a nucleotide sequencing-based genotyping method that uses highly variable intergenic spacers as typing markers. It is the most suitable genotyping procedure for evaluating the population structure of closely related strains of B. henselae (4). Previously, 50 MST genotypes from 201 B. henselae strains were phylogenetically organized into 4 lineages, and human strains mostly grouped within 2 of these lineages, Houston-1 and Marseille (5,6). Because genotypic data on B. henselae from Asian countries are limited, we applied MST to 56 human and cat specimens to determine the genotypic distribution and relationship of human and cat strains of B. henselae in Japan.
During 1997 through 2008, we collected 56 B. henselae specimens from western Japan, mainly from Yamaguchi prefecture; the specimens included 1 B. henselae isolate from a patient with endocarditis (7), 24 clinical specimens from CSD patients who had test results positive for B. henselae DNA, and 31 B. henselae isolates from domestic cats (8). The 24 clinical specimens included 5 lymph node specimens and 16 pus specimens from patients with typical CSD, 1 blood specimen from a patient with bacteremia, 1 liver specimen from a patient with hepatic granuloma, and 1 spleen specimen from a patient with splenic granuloma. Total genomic DNA was extracted from the specimens by using the QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany). B. henselae DNA was detected by using PCR with specific primers for the 16S–23S rRNA intergenic spacer (9) and the htrA gene (10), and the 16S rRNA genotype was confirmed by partial sequencing of the 16S rRNA gene (11).
In addition, previously described MST primers were used to amplify and sequence the 9 intergenic spacers in B. henselae DNA (6). Locus-specific PCR was performed for spacer S1; direct sequencing was unsuccessful because of an unusual number of variable number tandem repeats (VNTR) (8). Spacer sequences were assigned according to published data (5,6,8). For each of the 9 spacers (S1–S9), we identified 8, 2, 3, 4, 3, 2, 3, 2, and 3 genotypes, respectively (Table). Three novel spacer sequences were deposited in the DNA Data Bank of Japan with these accession numbers: AB558532, S2 genotype 9; AB558533, S4 genotype 7; and AB558534, S4 genotype 8. We identified 13 different MST genotypes among the 56 specimens (Table). Of these 13 MST genotypes, 7 were novel (types 51–57). Six MST genotypes belonged to human and cat strains, including 2 predominant MST genotypes (14 and 35). All MST data were deposited in the MST-Rick database (http://ifr48.timone.univ-mrs.fr/MST_BHenselae/mst).
Figure
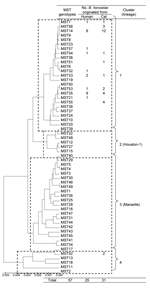
Figure. Phylogeny and clusters of multispacer typing (MST) genotypes of Bartonella henselae isolates from humans and cats, Japan, based on 9 concatenated intergenic spacer sequences in 57 MST genotypes. The unweighted pair-group...
Subsequently, we analyzed the phylogenetic relationships of the 7 novel MST genotypes identified in this study with the 50 previously identified genotypes. Multiple sequence alignment of the concatenated spacer sequences was performed by using ClustalW (www.ebi.ac.uk/clustalw). Finally, a phylogenetic tree was constructed by using the unweighted pair-group method with arithmetic mean (UPGMA) in MEGA4 (12). This phylogenetic tree is grouped into 4 clusters (Figure). Of the 13 MST genotypes identified in this study, 12 genotypes belonged to cluster 1, but one genotype (MST genotype 52) belonged to cluster 4.
This study showed that MST genotypes in Japan were mainly grouped into 1 lineage (cluster 1), which was composed of Asian and American strains of B. henselae, and that the genotypic distribution of human strains coincided with that of cat strains. Although only 1 human strain from the West Indies belonged to cluster 1 before this study (6), we discovered that all 25 of our human strains from Japan were grouped into cluster 1. These results demonstrate that human infections can be caused by B. henselae strains in cluster 1, which differed from clusters corresponding to the Houston-1 and Marseille type strains.
In a previous study, MST genotype 35 was the most common genotype in cluster 1, and 4/6 (67%) of Japanese cat strains belonged to this genotype (5). In this study, MST genotypes 14 (36%; 20/56) and 35 (23%; 13/56) were predominant genotypes. Additionally, most human strains (88%; 15/17) belonging to these genotypes were isolated from patients with typical CSD; 2 strains with MST genotype 35 were isolated from patients with endocarditis and bacteremia (Table).
The genotypic distribution of the human strains in this study differed from that reported by Li et al. (6) because their strains isolated in France were grouped under 2 lineages (Houston-1 and Marseille). However, we found that the lineages of human strains matched those of cat strains in each country. These results are consistent with the role of cats as the major reservoir of B. henselae (13).
In this study, we identified 2 cat strains that were classified into cluster 4. These strains belonged to 16S type II, which is rare in Japan (3). In previous MST studies, strains in cluster 4 were isolated from cats and belonged to 16S type II (5,6). Intriguingly, similar lineages consisting of 16S type II isolates from cats were observed in other genotyping studies involving the use of multilocus sequence typing (MLST) (14) and multiple locus variable number tandem repeat analysis (MLVA) (15). Thus, these lineages may be less pathogenic for humans. However, further studies are needed to investigate this hypothesis.
When we characterized the strains in this study by MLST we found that almost all of them shared the same sequence type as Houston-1 (8). In contrast, we identified 13 MST genotypes that belonged to different clusters than Houston-1. The lower resolving power of MLST is mostly likely due to sequence conservation in the 8 housekeeping genes selected for the method. MST has a higher resolving power because the spacers used in this method are more variable than MLST markers. As a result, MST is better suited for evaluating the population structure of closely related B. henselae strains.
We conclude that the MST genotypes in Japan are mainly grouped into cluster 1 and that the genotypic distribution of human strains coincides with that of cat strains. In Japan, human infections can be caused by B. henselae strains in cluster 1, distinct from clusters containing the Houston-1 and Marseille type strains. These results improve our understanding of the population structure of and geographic relationship between human and cat strains of B. henselae.
Dr Yanagihara is a medical technologist and research scientist in the Department of Basic Laboratory Sciences, Yamaguchi University Graduate School of Medicine. His research interests focus on B. henselae infections and their molecular epidemiology.
Acknowledgment
This work was supported by Grant-in-Aid for Young Scientists (B) No. 21790538 from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- Anderson BE, Neuman MA. Bartonella spp. as emerging human pathogens. Clin Microbiol Rev. 1997;10:203–19.PubMedGoogle Scholar
- Boulouis HJ, Chang CC, Henn JB, Kasten RW, Chomel BB. Factors associated with the rapid emergence of zoonotic Bartonella infections. Vet Res. 2005;36:383–410.PubMedGoogle Scholar
- Maruyama S, Nakamura Y, Kabeya H, Tanaka S, Sakai T, Katsube Y. Prevalence of Bartonella henselae, Bartonella clarridgeiae and the 16S rRNA gene types of Bartonella henselae among pet cats in Japan. J Vet Med Sci. 2000;62:273–9.PubMedGoogle Scholar
- Li W, Raoult D, Fournier PE. Bacterial strain typing in the genomic era. FEMS Microbiol Rev. 2009;33:892–916.PubMedGoogle Scholar
- Li W, Chomel BB, Maruyama S, Guptil L, Sander A, Raoult D, Multispacer typing to study the genotypic distribution of Bartonella henselae populations. J Clin Microbiol. 2006;44:2499–506.PubMedGoogle Scholar
- Li W, Raoult D, Fournier PE. Genetic diversity of Bartonella henselae in human infection detected with multispacer typing. Emerg Infect Dis. 2007;13:1178–83.PubMedGoogle Scholar
- Tsuneoka H, Yanagihara M, Otani S, Katayama Y, Fujinami H, Nagafuji H, A first Japanese case of Bartonella henselae induced endocarditis diagnosed by prolonged culture of a specimen from the excised valve. Diagn Microbiol Infect Dis. 2010;68:174–6.PubMedGoogle Scholar
- Yanagihara M, Tsuneoka H, Hoshide S, Ishido E, Umeda A, Tsukahara M, Molecular typing of Bartonella henselae DNA extracted from human clinical specimens and cat isolates in Japan. FEMS Immunol Med Microbiol. 2010;60:44–8.PubMedGoogle Scholar
- Jensen WA, Fall MZ, Rooney J, Kordick DL, Breitschwerdt EB. Rapid identification and differentiation of Bartonella species using a single-step PCR assay. J Clin Microbiol. 2000;38:1717–22.PubMedGoogle Scholar
- Anderson B, Sims K, Regnery R, Robinson L, Schmidt MJ, Goral S, Detection of Rochalimaea henselae DNA in specimens from cat scratch disease patients by PCR. J Clin Microbiol. 1994;32:942–8.PubMedGoogle Scholar
- Bergmans AM, Schellekens JF, van Embden JD, Schouls LM. Predominance of two Bartonella henselae variants among cat-scratch disease patients in the Netherlands. J Clin Microbiol. 1996;34:254–60.PubMedGoogle Scholar
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–9.PubMedGoogle Scholar
- Regnery R, Martin M, Olson J. Naturally occurring “Rochalimaea henselae” infection in domestic cat. Lancet. 1992;340:557–8.PubMedGoogle Scholar
- Arvand M, Feil EJ, Giladi M, Boulouis HJ, Viezens J. Multi-locus sequence typing of Bartonella henselae isolates from three continents reveals hypervirulent and feline-associated clones. PLoS ONE. 2007;2:e1346.PubMedGoogle Scholar
- Bouchouicha R, Durand B, Monteil M, Chomel BB, Berrich M, Arvand M, Molecular epidemiology of feline and human Bartonella henselae isolates. Emerg Infect Dis. 2009;15:813–6.PubMedGoogle Scholar
Figure
Table
Cite This ArticleTable of Contents – Volume 16, Number 12—December 2010
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Masashi Yanagihara, Department of Basic Laboratory Sciences, Yamaguchi University Graduate School of Medicine, 1-1-1 Minami-kogushi, Ube, Yamaguchi 755-8505, Japan
Top