Volume 16, Number 3—March 2010
Letter
Two Lineages of Dengue Virus Type 2, Brazil
Figure
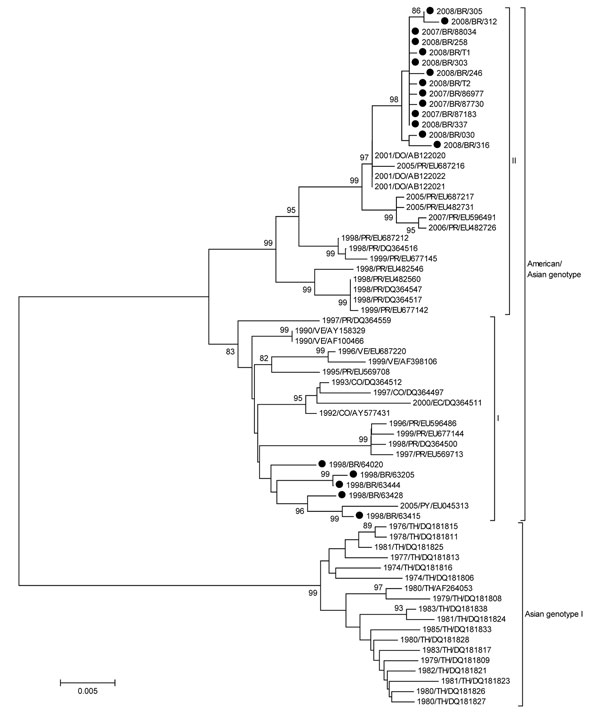
Figure. Neighbor-joining phylogenetic tree of 68 complete envelope (E) gene sequences of dengue virus type 2 (DENV-2). Only bootstrap values >80% are shown. DENV-2 sequences obtained from 21 patients infected during the 1990, 1998, and 2007–2008 epidemics were isolated from acute-phase cases. Viral RNA was extracted from 140 µL of serum or supernatant of infected C636 cell cultures by using the QIAamp Viral RNA Mini Kit (QIAGEN, Valencia, CA, USA), and cDNA synthesis was performed by using the ThermoScript reverse transcription–PCR System (Invitrogen, Carlsbad, CA, USA) according to the manufacturers’ recommendations. We obtained a fragment of 1,878 bp with the complete dengue virus envelope (E) gene sequence by seminested PCR using Taq Platinum DNA polymerase (Invitrogen). The PCR products were purified by using the Millipore (Billerica, MA, USA) purification column and sequenced by using BigDye Terminator version 3.1 Cycle Sequencing (Applied Biosystems, Foster City, CA, USA) and ABI3130 automated sequencer. Sequences of the E gene were compared with DENV-2 sequences of American/Asian genotype deposited in GenBank (www.ncbi.nlm.nih.gov). Strains of Asian genotype I served as the outgroup. All sequences were aligned by using ClustalX (www.ebi.ac.uk/clustalw), and phylogenetic analysis was performed by using MEGA 4.0 (www.megasoftware.net), according to the Tamura-Nei model. First, neighbor-joining trees were constructed by using a 798 pb of the at 5′ region of the E gene and later trees were constructed with sequences from the 1998, and 2007–2008 epidemics by using the complete E gene sequence (1,485 bp). The reliability of the inferred phylogenetic tree was estimated by the bootstrap method, with 1,000 replications. Horizontal branch lengths are drawn to scale, and the tree was rooted by using the Asian genotype, which always appears as the most divergent. Scale bar represents percentage of genetic distance. The names of DENV-2 isolates include reference to year of isolation and country of origin: BR, Brazil; CO, Colombia; DO, Dominican Republic; EC, Ecuador; PR, Puerto Rico; PY, Paraguay; TH, Thailand; VE, Venezuela. GenBank accession nos: 1990/BR/39537 (GQ368156); 1990/BR/40044 (GQ368157); 1998/BR/63415 (GQ368158); 1998/BR/63428 (GQ368159); 1998/BR/63205 (GQ368160); 2007/BR/86977 (GQ368161); 2007/BR/87183 (GQ368162); 2007/BR/87730 (GQ368163); 2007/BR/88034 (GQ368164); 2008/BR/030 (GQ368165); 2008/BR/246 (GQ368166); 2008/BR/258 (GQ368167); 2008/BR/303 (GQ368168); 2008/BR/305 (GQ368169); 2008/BR/312 (GQ368170); 2008/BR/316–08 (GQ368171); 2008/BR/337–08 (GQ368172); 1998/BR/63444 (GQ368173); 1998/BR/64020 (GQ368174); 2008/BR/T2 (GQ368175); 2008/BR/T1 (GQ368176).