Volume 16, Number 6—June 2010
Research
Evolution of Northeastern and Midwestern Borrelia burgdorferi, United States
Cite This Article
Citation for Media
Abstract
The per capita incidence of human Lyme disease in the northeastern United States is more than twice that in the Midwest. However, the prevalence of Borrelia burgdorferi, the bacterium that causes Lyme disease, in the tick vector is nearly identical in the 2 regions. The disparity in human Lyme disease incidence may result from a disparity in the human invasiveness of the bacteria in the Northeast and Midwest caused by fundamentally different evolutionary histories. B. burgdorferi populations in the Northeast and Midwest are geographically isolated, enabling evolutionary divergence in human invasiveness. However, we found that B. burgdorferi populations in the Northeast and Midwest shared a recent common ancestor, which suggests that substantial evolutionary divergence in human invasiveness has not occurred. We propose that differences in either animal ecology or human behavior are the root cause of the differences in human incidence between the 2 regions.
Lyme disease, caused by the bacterium Borrelia burgdorferi, is the most common vector-borne disease in the United States (1). B. burgdorferi is transmitted to humans during the blood meal of an infected Ixodes tick, predominantly Ixodes scapularis in North America (2). The prevalence and density of B. burgdorferi–infected I. scapularis ticks are nearly identical in the northeastern and midwestern United States, the regions with the highest incidence of Lyme disease in humans (3–6); however, the number of human Lyme disease cases reported in the Northeast and Midwest is not (1). The overwhelming majority of Lyme disease cases in the United States are reported from the Northeast (82%), followed distantly by the Midwest (9%) (1). Similarly, per capita Lyme disease incidence is >2× greater in the Northeast than the Midwest. We address the hypothesis that B. burgdorferi populations in the Northeast and Midwest have fundamentally different evolutionary histories, which may result in differing degrees of human invasiveness.
The evolutionary and demographic histories of B. burgdorferi in the Northeast have been intensively studied. The effective population size of northeastern B. burgdorferi is small because of its recent colonization of the northern United States and its life-history strategy (7,8). The strikingly impoverished neutral genetic diversity and high linkage disequilibrium within B. burgdorferi populations likely result from small effective population sizes (9–11). Genetic loci are found in perfect or near-perfect association in B. burgdorferi in the Northeast (9–12). Strong linkage disequilibrium among genetic loci can result from several evolutionary and ecologic forces in addition to small population size (drift), such as lack of recombination machinery or limited opportunity for gene exchange (13). Genetically diverse strains of B. burgdorferi often are found within the same tick or same vertebrate host, suggesting ample opportunity for genetic exchange (4,14). Evidence is strong that recombination occurs within a genomic lineage of B. burgdorferi (15–17). Thus, B. burgdorferi has the opportunity and the recombination system needed for genetic exchange. A historically small effective population size is a parsimonious explanation for the low neutral genetic diversity and strong linkage disequilibrium.
The evolutionary and demographic histories of B. burgdorferi in the Midwest are comparatively understudied. On the coarse scale, the evolutionary history and ecology of B. burgdorferi in the Northeast and Midwest appear similar. Both regions have oak-maple–dominated forests ideal for deer and the small mammals that maintain the I. scapularis and B. burgdorferi populations, were under the Pleistocene ice sheet, and were recently colonized by B. burgdorferi. The differences accounting for a lower Lyme disease incidence in the Midwest than the Northeast are not clear but most likely can be found on a finer scale in the evolutionary history of B. burgdorferi (this study), the tick vector (8), or human exposure.
The differences accounting for a lower Lyme disease incidence in the Midwest than the Northeast are not clear but are likely to be found on a finer evolutionary or ecologic scale. A recent report suggested that a lower proportion of the 16S–23S rRNA intergenic spacer (IGS) type 1 (RST-1) allele in midwestern B. burgdorferi populations, an allele associated with human invasiveness in the northeastern United States, may account for the differences in human Lyme disease incidence (8). However, the 16S–23S rRNA IGS does not directly influence B. burgdorferi invasiveness; it is in linkage disequilibrium with a gene of major effect in the Northeast. The linkage patterns in midwestern B. burgdorferi are as yet unstudied. A fundamentally different evolutionary history would result in divergent linkage disequilibrium patterns between the Northeast and the Midwest and potentially result in differing degrees of human invasiveness associated with alleles at the 16S–23S rRNA spacer.
For this study, we used a phylogenetic framework to analyze the evolutionary and demographic histories of B. burgdorferi in the midwestern and northeastern United States (18). A previous multilocus sequencing typing study of 4 B. burgdorferi loci (outer surface protein C [ospC], 16S–23S rRNA (rrs-rrl-A), ospA, and outer membrane protein [p66]) from strains isolated in the Northeast identified 9 distinct lineages with complete linkage among alleles at the 4 loci (10). That study analyzed statistical associations of haplotypes at each locus without regard to the underlying evolutionary relationships of sequences. In this study, we analyzed ospC, rrs-rrlA, ospB, and ospA (p66 contains little evolutionary information [10]) from midwestern and northeastern populations in a phylogenetic framework to investigate the shared and vicariant evolutionary and demographic histories of B. burgdorferi from geographically isolated regions.
B. burgdorferi Isolates
All isolates were derived from skin biopsy specimens of 47 adult patients who had erythema migrans, at the Marshfield Clinic in central Wisconsin during 1995–2001. Specimens were collected and cultured as described elsewhere (19).
DNA Extraction and Amplification
The DNA sequences of ospA, ospB, rrs-rrlA, and ospC were determined for use in our phylogenetic analysis. Cultivated isolates were harvested by centrifugation at 5,000 × g for 15 min, resuspended in sterile water, and lysed by boiling for 5 min. PCR conditions are described by Bunikis et al. (10) for ospA and rrs-rrlA, by Caporale and Kocher (20) for ospB, and by Brisson and Dykhuizen (4) for ospC. Negative controls were included for DNA extraction and PCR procedures to monitor for contamination. Amplified PCR products were sequenced in both directions. ospC products were subject to the reverse-line blot procedure as described previously (4,7). ospC amplicons that could not be classified to a major allelic group were sequenced.
Analyses
All analyses included sequence data collected in this study and reported in Bunikis et al. (10). DNA sequences were aligned using the Clustal X algorithm (21) with default settings, and alignments were refined manually where necessary. Descriptive statistics (π, synonymous and nonsynonymous polymorphisms) were determined using DNAsp (ver. 4.50.3) (22). Two tests for recombination within genes (Sawyer test and maximum χ2 test) and a test for allelic association (index of association [IA]) were performed in START2 (23). Sawyer runs test compares pairs of alleles to determine whether regions of the sequence space have more consecutive identical polymorphisms (runs) than expected by chance (24). The maximum χ2 test uses the distribution of polymorphic sites to identify potential recombination events between pairs of alleles (25). The IA assesses the extent of association between loci using allele frequencies without regard to the underlying sequences (26).
Phylogenetic reconstruction was performed using Bayesian (MrBayes; http://mrbayes.csit.fsu.edu) and maximum likelihood (PAUP*, version 4.0; http://paup.csit.fsu.edu) approaches. Trees were constructed from the sequence data from each gene individually and from combinations of genes. IGS, ospA, and ospB provide reliable data for phylogenetic inference and can be easily compared with previous analyses (10). ospC DNA sequences provide information about disease invasiveness (27,28) but were not used in phylogenetic reconstruction because the gene tree topology is star-shaped with short and unsupported internal branches, long terminal branches, and many polytomies. This topology provides no information about the evolutionary history of this gene or the relationships among alleles. When information about ospC was included in the data set, it was included as a heavily weighted morphologic character in MrBayes, not as DNA sequence data. The morphologic data constrain the isolates with the same ospC allele cluster but do not affect the topology of the internal branches of the tree, which depend only on the DNA sequences of the other genes in the data set. For each data set, we used Modeltest 3.4 (29) to select the appropriate model of molecular evolution. We used this model to find the Bayesian tree and the maximum likelihood tree.
We used the Shimodaira-Hasegawa (SH) test as implemented in PAUP to test for differences in evolutionary histories among genes using the resampling of estimated log-likelihoods approximation with 1,000 bootstrap replicates (18). This method tests for significant differences in the likelihood of several given tree topologies given a data set. The likely tree topologies produced by each locus analyzed in this study were used as the set of topologies. If no true difference exists, each data set is equally likely to produce any of the given tree topologies, and all genes in this study share similar evolutionary histories. Alternatively, a significantly lower likelihood suggests that recombination has occurred between the genes in the data set and the gene used to create the lower-likelihood tree topology.
Sequence Diversity
B. burgdorferi isolates were cultured from primary or secondary skin legions of 47 adults visiting the Marshfield Clinic during 1995–2001. The sequences at the IGS, ospA, ospB, and ospC loci were determined from 39, 31, 44, and 44 strains of these B. burgdorferi isolates, respectively. Erythema migrans represented the bulk of the diversity found in tick samples (27) and provided a representative sample of B. burgdorferi bacteria in the Midwest. Furthermore, we found all of the IGS types found by Bunikis et al. (10), suggesting the sample from the Midwest was not biased in terms of the types found. ospA showed little evolutionarily information and was not determined from 14 of the isolates. One sequence was found at each locus in each culture.
Nucleotide diversity varied considerably among loci (Table 1). We found 47 polymorphic sites in the IGS sequence sample, resulting in 16 unique haplotypes (Technical Appendix). Six haplotypes were identical to haplotypes previously found in the Northeast, and 38 of the 47 polymorphic sites found in the Northeast were present in this sample from the Midwest, supporting a recent shared ancestry of B. burgdorferi from the 2 populations (Technical Appendix). The 10 unique haplotypes in the Midwest and 9 unique polymorphic sites suggest some isolation by distance and an emerging evolutionary divergence.
ospA and ospB are considerably less diverse than IGS (Table 1). Nevertheless, there are 14 unique ospA haplotypes and 14 unique ospB haplotypes in this sample (Technical Appendix). Nine of the ospA haplotypes in the samples from the Midwest also are found in B. burgdorferi in the Northeast. The diversity of ospB has not been examined in the Northeast. Amino acid sequence evolution in ospA and ospB appears to be constrained because synonymous changes greatly exceed nonsynonymous changes (dNospA/dSospA = 0.334; dNospB/dSospB = 0.303).
The mean pairwise diversity at ospC was much greater than at the other loci analyzed (π = 0.207). Sixty-three changes resulted in amino acid substitutions. However, we found no evidence of directional selection (dN/dS = 0.591). Eighteen major groups of alleles, which differ by >8% in sequence, are represented in the isolates from the Midwest (Appendix Table). Fourteen of the 18 major group alleles are identical to those found in the Northeast, although types L and O are rare in the Northeast. We found 4 novel ospC major group alleles (W, V, Y, Z) in these samples (Appendix Table). ospC major groups W, V, and X also were detected in tick populations in the Midwest (D. Brisson, unpub. data), although we did not see type X in this set of human isolates. Strains with ospC major groups I, J, and T, which are present in tick and human samples in the Northeast, were absent in this sample from the Midwest.
Phylogenies
Figure
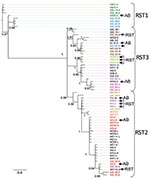
Figure. Phylogeny of Borrelia burgdorferi isolates in the northeastern and midwestern United States based on intergenic spacer (IGS) sequence. operational taxanomic unit names beginning with IGS were isolated in the northeastern United...
We used Bayesian and likelihood algorithms to reconstruct phylogenies for each gene. The 2 approaches resulted in identical topologies for each gene except ospC. The IGS gene tree forms a strongly supported phylogeny with most nodes bifurcating (Figure). Polychotomies occur only at sequences that are identical or differed by 1 nucleotide. The phylogeny generally supports the previously described divisions of IGS sequences into the 3 restriction fragment length subgroups, arbitrarily called RST 1, RST2, and RST3 (31). However, the diverse RST3 group is not monophyletic. The IGS sequences from strains in the Northeast and Midwest are interdigitated, suggesting a recent shared history.
The gene trees for ospA and ospB individually have many weakly supported nodes and polychotomies because of the limited number of phylogenetically informative sites. The phylogeny reconstructed from the ospA/ospB operon (concatenated data set) result in much stronger support for internal nodes. Nevertheless, the ospAB phylogeny contains 1 polychotomy. Concatenating the data sets for ospA and ospB sequences is appropriate because these loci lie contiguously in a single operon, and little evidence exists for recombination either within or between these genes (see below). The northeastern and midwestern strains were interdigitated on the ospA, ospB, and ospAB trees, which suggests a recent, shared population history.
Analysis of the ospC data set yielded a phylogeny with short internal branches, long terminal branches, and no supported internal nodes, commonly referred to as a star phylogeny (10,13). ospC thus appeared to have evolved by recombination (9,32). Phylogenies of highly recombining loci are poorly supported because the evolutionary history differed in different segments of the allele (33).
Intragenic Recombination
We found little evidence of recombination within IGS, ospA, or ospB in the Midwest data set or the combined northeastern/midwestern data set (Table 1). Sawyer runs test found no runs of polymorphisms that are significantly incongruous within the sequence. Maynard Smith’s maximum χ2 test found 1 recombination event within the IGS sequence at nt 107 (position compared with B31 type strain) in both data sets. We found no evidence for recombination within ospA and only 1 potential site of recombination at the extreme 3′ end of ospB. Neither maximum χ2 nor Sawyer test found evidence for recombination between ospA and ospB in the ospAB operonic data set. However, ospA haplotypes are often associated with multiple ospB haplotypes suggesting recombination or independent evolution (Technical Appendix). Intragenic recombination was evident within ospC sequences. Sawyer test identified 4 runs of polymorphisms that appear to be transferred from another ospC major allelic group. Maynard Smith maximum χ2 test found evidence of recombination at 5 locations along the sequence (Table 1).
Intergenic Recombination
We assessed intergenic recombination using the IA and the SH test. The index of association detects nonrandom associations of haplotypes among genes but does not account for sequence variation or relatedness. The IA indicated that all loci examined in this study were significantly nonrandomly associated, i.e., in linkage disequilibrium (p<0.0001) (Table 1).
The SH test uses phylogenetic information to identify horizontal transfer events by comparing the evolutionary histories of genes as represented by the gene-tree topologies. The likelihood of the IGS sequence data resulting in the ospAB tree topology is significantly less likely than these data resulting in the IGS topology (Table 2). That is, the evolutionary history of the IGS locus is not congruent with the ospAB evolutionary history, providing evidence of lateral gene transfers. Strains found in different strongly supported clades on the ospAB and the IGS trees are identified in the Figure (p<0.0001).
The phylogenies reconstructed from midwestern ospAB sequences were compared with ospAB phylogenies that constrain strains with the same ospC allele to cluster (ospABC tree). We found no statistical support for recombination between ospC and ospAB (p = 0.313). However, 4 ospC type K strains (W9, W7, MC2, KR3) are separated from the other K strains by supported nodes in the ospAB tree, and 1 ospC type F strain (MC102) is separated from the other ospC type F strains. We also found no evidence of recombination between ospA and ospC or ospB and ospC (p>0.1). However, the ospA tree is nearly significantly more likely than the ospAC tree when strains from the Northeast are included in the analysis (p = 0.08).
Several instances of recombination are evident between IGS and ospC. The differences in evolutionary history were observable by using the midwestern IGS data set alone (p = 0.006) and the combined midwestern/northeastern IGS data set (p<0.001). These analyses provide evidence that linkage patterns in B. burgdorferi from the Northeast differ from that of the linkage patterns in B. burgdorferi from the Midwest.
The divergence in human Lyme disease incidence between the Northeast and Midwest does not result from independent evolution of human invasiveness because of geographic isolation. Although pathogen populations in the Midwest appear geographically isolated from those in the Northeast (6), evolutionary and demographic analyses indicate that they share a recent common ancestor. Both populations have little standing genetic variation, as indicated by the limited number of polymorphic sites, suggesting small effective population sizes and similar life-history strategies. The combination of linked alleles also is similar in both regions, supporting the recent shared ancestor hypothesis. B. burgdorferi strains isolated in the Midwest are interleaved with northeastern strains on phylogenetic trees evincing their close evolutionary relationship. However, there is some genetic divergence and differing linkage groups between the regions, intimating that gene flow is limited between these populations, allowing them to differentiate. The recent common ancestor in the northeastern and midwestern B. burgdorferi and limited genetic divergence suggests that human Lyme disease incidence cannot be explained by fundamentally different evolutionary histories resulting in differing degrees of human infectiousness.
None of the loci investigated show substantial genetic divergence between regions, suggesting a recent common ancestor and similar phenotypes. Northeastern haplotypes are interleaved with midwestern haplotypes such that the time to coalescence of alleles within a region is equivalent to the time to coalescence for alleles from both regions (Figure). These data suggest that northeastern and midwestern strains have a recent common ancestor. The limited genetic diversity in B. burgdorferi in the Midwest and Northeast (Technical Appendix) suggests that the populations have retained the life-history strategy of their common ancestor (10,12). However, isolation by distance and subsequent divergence resulted in unique alleles in each region.
The IGS gene tree reconstructed from midwestern and northeastern data broadly supports the RST system described using northeastern populations (31). RST types 1 and 2 form strongly supported monophyletic groups. RST3 is polyphyletic and should be split into 3 groups as defined by the strongly supported clades (Figure). Supporting this suggestion, RST3 is diverse genetically and phenotypically (34). Interestingly, this division would separate ospC major group I bearing strains, a particularly invasive group in humans, from the other RST3 strains that rarely cause disseminated infections in humans (27,28,30).
The ospC data support the hypothesis that the strains from the Northeast and Midwest have a common ancestor but are currently isolated and have begun to diverge. Most ospC major groups are found in both regions. Given the genetic distance between major group alleles, the exact set of alleles is unlikely to have occurred twice independently. Additionally, the linkage relationships between ospC alleles and IGS alleles are similar in both regions. Both lines of evidence suggest that most of the diversity at ospC originated before the northeastern and midwestern populations diverged. Differences in invasiveness between B. burgdorferi in the Northeast and Midwest do not result from fundamentally different evolutionary histories.
Four novel ospC major group alleles appear to be unique to the Midwest (Table 2). In addition to these novel ospC major groups, a type C–like allele appears to have been generated independently in the Midwest and the Northeast. The group C allele in the Midwest shares 96.8% similarity with the group C allele in the Northeast. Whether the unique ospC major group alleles were generated recently in only 1 region or whether they were shared in the ancestral population and subsequently lost in only 1 region is not clear.
B. burgdorferi lineages exchange DNA, contrary to previous reports (10,11,13). We found at least some evidence of recombination between all genetic loci examined; even the ospAB operon has several homoplasious mutations, suggesting potential recombination (Technical Appendix). However, recombination between ospA and ospB is not statistically supported and may have arisen from recurrent mutation (Appendix Table). Despite evidence for recombination, the linkage relationships are similar in the Northeast and the Midwest, supporting the recent common ancestry of these populations.
Recombination is more apparent in Lyme disease foci in the Midwest than in the Northeast (Figure; Appendix Table). This is likely to be caused by neutral divergence of linkage patterns resulting from small effective population sizes in both regions coupled with gene flow from the Northeast to the Midwest but not in the other direction. Small effective population sizes eliminate most of the linkage combinations in each region such that they are in perfect linkage disequilibrium. Gene flow from the northeastern population then introduces linkage pattern variation into the midwestern population. Linkage patterns unique to the Midwest are absent from the Northeast, suggesting that gene flow from the Midwest to the Northeast is rare.
B. burgdorferi in the Northeast and Midwest share a remarkably similar evolutionary history. Independent evolution of human invasiveness in the 2 regions does not explain the lower human Lyme disease incidence in the Midwest. Other potential causes for the differences in human Lyme disease incidence include differences in human exposure to B. burgdorferi–infected ticks and ecologic differences in the reservoir host community. Lyme disease typically is contracted peridomestically in the Northeast (35), but similar studies reporting the peridomestic acquisition of B. burgdorferi have not been reported from the Midwest. Human risk for exposure to Lyme disease also may be exaggerated in the Northeast because of the immense suburban populations around the major metropolitan areas. Current ecologic conditions yielding differences in the composition of reservoir host species could alter the prevalence of B. burgdorferi lineages that are particularly invasive in humans (27,28,30,36,37). For example, midwestern ticks are rarely infected with ospC genotypes A or B (RST I) (3), 2 of the 4 genotypes that are common in the Northeast and regularly cause human Lyme disease (27,28,30).
Dr Brisson is an assistant professor in the biology department at the University of Pennsylvania. His primary research interests focus on the ecology and evolution of infectious diseases.
Acknowledgment
This work was supported by National Institute of Health grants AI076342 (D.B.) and GM31912 (D.E.D) and Centers for Diseases Control and Prevention grant U01CK000170 (D.B.).
References
- Bacon RM, Kugeler KJ, Mead PS. Surveillance for Lyme disease—United States, 1992–2006. MMWR Surveill Summ. 2008;57:1–9.PubMedGoogle Scholar
- Burgdorfer W, Barbour AG, Hayes SF, Benach JL, Grunwaldt E, Davis JP. Lyme disease—a tick-borne spirochetosis. Science. 1982;216:1317–9. DOIPubMedGoogle Scholar
- Gatewood AG, Liebman KA, Vourc’h G, Bunikis J, Hamer SA, Cortinas R, Climate and tick seasonality are predictors of Borrelia burgdorferi genotype distribution. Appl Environ Microbiol. 2009;75:2476–83. DOIPubMedGoogle Scholar
- Brisson D, Dykhuizen DE. ospC diversity in Borrelia burgdorferi: different hosts are different niches. Genetics. 2004;168:713–22. DOIPubMedGoogle Scholar
- Caporale DA, Johnson CM, Millard BJ. Presence of Borrelia burgdorferi (Spirochaetales: Spirochaetaceae) in southern Kettle Moraine State Forest, Wisconsin, and characterization of strain W97F51. J Med Entomol. 2005;42:457–72. DOIPubMedGoogle Scholar
- Diuk-Wasser MA, Gatewood AG, Cortinas MR, Yaremych-Hamer S, Tsao J, Kitron U, Spatiotemporal patterns of host-seeking Ixodes scapularis nymphs (Acari: Ixodidae) in the United States. J Med Entomol. 2006;43:166–76. DOIPubMedGoogle Scholar
- Qiu WG, Dykhuizen DE, Acosta MS, Luft BJ. Geographic uniformity of the Lyme disease spirochete (Borrelia burgdorferi) and its shared history with tick vector (Ixodes scapularis) in the northeastern United States. Genetics. 2002;160:833–49.PubMedGoogle Scholar
- Humphrey PT, Caporale DA, Brisson D. Uncoordinated biogeography of the Lyme disease pathogen, Borrelia burgdorferi, and its tick vector, Ixodes scapularis. Evolution. 2010. In press. DOIPubMedGoogle Scholar
- Qiu WG, Schutzer SE, Bruno JF, Attie O, Xu Y, Dunn JJ, Genetic exchange and plasmid transfers in Borrelia burgdorferi sensu stricto revealed by three-way genome comparisons and multilocus sequence typing. Proc Natl Acad Sci U S A. 2004;101:14150–5. DOIPubMedGoogle Scholar
- Bunikis J, Tsao J, Berglund J, Fish D, Barbour AG. Sequence typing reveals extensive strain diversity of the Lyme borreliosis agents Borrelia burgdorferi in North America and Borrelia afzelii in Europe. Microbiology. 2004;150:1741–55. DOIPubMedGoogle Scholar
- Dykhuizen DE, Polin DS, Dunn JJ, Wilske B, Preac-Mursic V, Dattwyler RJ, Borrelia burgdorferi is clonal: implications for taxonomy and vaccine development. Proc Natl Acad Sci U S A. 1993;90:10163–7. DOIPubMedGoogle Scholar
- Attie O, Bruno JF, Xu Y, Qiu D, Luft BJ, Qiu WG. Co-evolution of the outer surface protein C gene (ospC) and intraspecific lineages of Borrelia burgdorferi sensu stricto in the northeastern United States. Infect Genet Evol. 2007;7:1–12. DOIPubMedGoogle Scholar
- Dykhuizen DE, Baranton G. The implications of a low rate of horizontal transfer in Borrelia. Trends Microbiol. 2001;9:344–50. DOIPubMedGoogle Scholar
- Guttman DS, Wang PW, Wang IN, Bosler EM, Luft BJ, Dykhuizen DE. Multiple infections of Ixodes scapularis ticks by Borrelia burgdorferi as revealed by single-strand conformation polymorphism analysis. J Clin Microbiol. 1996;34:652–6.PubMedGoogle Scholar
- Casjens S, Palmer N, van Vugt R, Huang WM, Stevenson B, Rosa P, A bacterial genome in flux: the twelve linear and nine circular extrachromosomal DNAs in an infectious isolate of the Lyme disease spirochete Borrelia burgdorferi. Mol Microbiol. 2000;35:490–516. DOIPubMedGoogle Scholar
- Stevenson B, Miller JC. Intra- and interbacterial genetic exchange of Lyme disease spirochete erp genes generates sequence identity amidst diversity. J Mol Evol. 2003;57:309–24. DOIPubMedGoogle Scholar
- Zhang J-R, Norris SJ. Genetic variation of the Borrelia burgdorferi gene vlsE involves cassette-specific, segmental gene conversion. Infect Immun. 1998;66:3698–704.PubMedGoogle Scholar
- Shimodaira H, Hasegawa M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol. 1999;16:1114–6.
- Wormser GP, Liveris D, Nowakowski J, Nadelman RB, Cavaliere LF, McKenna D, Association of specific subtypes of Borrelia burgdorferi with hematogenous dissemination in early Lyme disease. J Infect Dis. 1999;180:720–5. DOIPubMedGoogle Scholar
- Caporale DA, Kocher TD. Sequence variation in the outer-surface-protein genes of Borrelia burgdorferi. Mol Biol Evol. 1994;11:51–64.PubMedGoogle Scholar
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80. DOIPubMedGoogle Scholar
- Rozas J, Rozas R. DnaSP version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics. 1999;15:174–5. DOIPubMedGoogle Scholar
- Jolley KA, Feil EJ, Chan MS, Maiden MC. Sequence type analysis and recombinational tests (START). Bioinformatics. 2001;17:1230–1. DOIPubMedGoogle Scholar
- Sawyer S. Statistical tests for detecting gene conversion. Mol Biol Evol. 1989;6:526–38.PubMedGoogle Scholar
- Smith JM. Analyzing the mosaic structure of genes. J Mol Evol. 1992;34:126–9. DOIPubMedGoogle Scholar
- Smith JM, Smith NH, O’Rourke M, Spratt BG. How clonal are bacteria? Proc Natl Acad Sci U S A. 1993;90:4384–8. DOIPubMedGoogle Scholar
- Seinost G, Dykhuizen DE, Dattwyler RJ, Golde WT, Dunn JJ, Wang IN, Four clones of Borrelia burgdorferi sensu stricto cause invasive infection in humans. Infect Immun. 1999;67:3518–24.PubMedGoogle Scholar
- Dykhuizen DE, Brisson D, Sandigursky S, Wormser GP, Nowakowski J, Nadelman RB, The propensity of different Borrelia burgdorferi sensu stricto genotypes to cause disseminated infections in humans. Am J Trop Med Hyg. 2008;78:806–10.PubMedGoogle Scholar
- Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–8. DOIPubMedGoogle Scholar
- Wormser GP, Brisson D, Liveris D, Hanincová K, Sandigursky S, Nowakowski J, Borrelia burgdorferi genotype predicts the capacity for hematogenous dissemination during early Lyme disease. J Infect Dis. 2008;198:1358–64. DOIPubMedGoogle Scholar
- Liveris D, Wormser GP, Nowakowski J, Nadelman R, Bittker S, Cooper D, Molecular typing of Borrelia burgdorferi from Lyme disease patients by PCR-restriction fragment length polymorphism analysis. J Clin Microbiol. 1996;34:1306–9.PubMedGoogle Scholar
- Livey I, Gibbs CP, Schuster R, Dorner F. Evidence for lateral transfer and recombination in OspC variation in Lyme disease Borrelia. Mol Microbiol. 1995;18:257–69. DOIPubMedGoogle Scholar
- Ragan MA. Detection of lateral gene transfer among microbial genomes. Curr Opin Genet Dev. 2001;11:620–6. DOIPubMedGoogle Scholar
- Wang G, Ojaimi C, Wu H, Saksenberg V, Iyer R, Liveris D, Disease severity in a murine model of Lyme borreliosis is associated with the genotype of the infecting Borrelia burgdorferi sensu stricto strain. J Infect Dis. 2002;186:782–91. DOIPubMedGoogle Scholar
- Cromley EK, Cartter ML, Mrozinski RD, Ertel SH. Residential setting as a risk factor for Lyme disease in a hyperendemic region. Am J Epidemiol. 1998;147:472–7.PubMedGoogle Scholar
- Brisson D, Dykhuizen DE. A modest model explains the distribution and abundance of Borrelia burgdorferi strains. Am J Trop Med Hyg. 2006;74:615–22.PubMedGoogle Scholar
- Brisson D, Dykhuizen DE, Ostfeld RS. Conspicuous impacts of inconspicuous hosts on the Lyme disease epidemic. Proc Biol Sci. 2008;275:227–35. DOIPubMedGoogle Scholar
Figure
Tables
Cite This ArticleTable of Contents – Volume 16, Number 6—June 2010
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Dustin Brisson, Department of Biology, University of Pennsylvania, Leidy Laboratories 209, 433 S University Ave, Philadelphia, PA 19104-6018, USA
Top