Volume 16, Number 6—June 2010
Letter
Toscana Virus Infection Imported from Elba into Switzerland
Figure
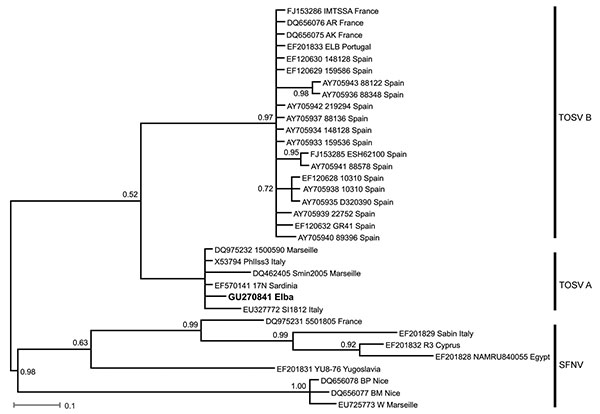
Figure. Bayesian phylogenetic tree of Toscana virus (TOSV) and Sandfly fever Naples virus (SFNV) strains. For each sequence used, GenBank accession number, strain designation, and strain origin are shown. Phylognetic analysis was performed by using MrBayes 3.0 program (4) with a general time reversible substitution model. Substitution rates were assumed to follow a gamma plus invariants distribution. Three heated chains and a single cold chain were used in all Markov Chain Monte Carlo analyses, which were run for 1,000,000 generations, sampling 1 tree every 100 generations. Trees obtained before convergent and stable likelihood values were discarded (i.e., a 2,500 tree burn-in). Four independent runs, each started from different, randomly chosen trees, were performed to assess convergence. Posterior probabilities for nodes were assembled from all post burn-in trees (i.e., 30,004 trees per analysis). Posterior probabilities are shown on each node. Scale bar indicates nucleotide substitutions per site. The newly described TOSV sequence from Elba is shown in boldface.
References
- Charrel RN, Gallian P, Navarro-Mari JM, Nicoletti L, Papa A, Sánchez-Seco MP, Emergence of Toscana virus in Europe. Emerg Infect Dis. 2005;11:1657–63.PubMedGoogle Scholar
- Dionisio D, Esperti F, Vivarelli A, Valassina M. Epidemiological, clinical and laboratory aspects of sandfly fever. Curr Opin Infect Dis. 2003;16:383–8. DOIPubMedGoogle Scholar
- Weidmann M, Sanchez-Seco MP, Sall AA, Ly PO, Thiongane Y, Lô MM, Rapid detection of important human pathogenic Phleboviruses. J Clin Virol. 2008;41:138–42. DOIPubMedGoogle Scholar
- Ronquist F, Huelsenbeck JP. Mrbayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–4. DOIPubMedGoogle Scholar
- Dobler G, Treib J, Haass A, Frösner G, Woesner R, Schimrigk K. Toscana virus infection in German travellers returning from the Mediterranean. Infection. 1997;25:325. DOIPubMedGoogle Scholar
- Sonderegger B, Hachler H, Dobler G, Frei M. Imported aseptic meningitis due to Toscana virus acquired on the island of Elba, Italy, August 2008. Euro Surveill. 2009;14:pii=19079.
- Charrel RN, Moureau G, Temmam S, Izri A, Marty P, Parola P, Massilia virus, a novel Phlebovirus (Bunyaviridae) isolated from sandflies in the Mediterranean. Vector Borne Zoonotic Dis. 2009;9:519–30. DOIPubMedGoogle Scholar
- Collao X, Palacios G, Sanbonmatsu-Gámez S, Pérez-Ruiz M, Negredo AI, Navarro-Marí JM, Genetic diversity of Toscana virus. Emerg Infect Dis. 2009;15:574–7. DOIPubMedGoogle Scholar
- Towards regional economic development 2006/2010. Tourism and innovations objectives and instruments [in Italian]. 2006 [cited 2010 Apr 1]. http://www.provincia.livorno.it/economia/turismo/forumturismo2006/FormatLivorno4aprile2006.pdf