Volume 17, Number 10—October 2011
Dispatch
Global Distribution of Shigella sonnei Clones
Cite This Article
Citation for Media
Abstract
To investigate global epidemiology of Shigella sonnei, we performed multilocus variable number tandem repeat analysis of 1,672 isolates obtained since 1943 from 50 countries on 5 continents and the Pacific region. Three major clonal groups were identified; 2 were globally spread. Type 18 and its derivatives have circulated worldwide in recent decades.
Shigella sonnei is the most commonly isolated species among the 4 Shigella species in industrialized countries (1,2). Transmission of S. sonnei across geographic boundaries is frequently linked to international travel and cross-border food trade (3,4). S. sonnei is a monomorphic organism and therefore requires a highly discriminative sequence-based method for investigating its clonal structure and the geographic distribution of clones.
A total of 26 variable number tandem repeats (VNTRs) have been used to type S. sonnei isolates (5). Because VNTRs have a wide range of variability, they are useful markers for investigating clonal relationships among strains that have evolved over different times (6). In this study, we analyzed 1,672 S. sonnei isolates obtained since 1943 from 50 countries on 5 continents (Africa, Asia, Europe, North America, and South America) and the Pacific region by multilocus VNTR analysis (MLVA) to investigate the global epidemiology of S. sonnei.
Isolates were obtained from 50 countries on 5 continents and the Pacific region (Table A1). Of these isolates, 31 were obtained during 1943–1983 (from Cameroon, Denmark, France, Senegal, and Sweden) and 1,641 were obtained during 1994–2008 from 48 countries. Isolates were lyophilized, kept in stab culture medium, or stored in 15%–20% glycerol at –75°C for long-term storage. The isolates were not repeatedly subcultured before this study. MLVA26, an MLVA assay based on analysis of 26 VNTRs, classified the 1,672 isolates into 620 MLVA26 types. With only 2 exceptions, no common MLVA26 type was shared among isolates from different countries. One isolate from Malaysia (1999) and 2 isolates from Vietnam (2006) shared a common MLVA26 type, and an isolate from Chad and an isolate from France (both isolated in 2007) shared a common MVLA26 type.
The high resolving power of MLVA is primarily caused by highly diverse VNTRs (6). Clustering analysis of the MLVA26 types using a minimum spanning tree (MST) algorithm grouped the 1,672 isolates into 3 large clusters (A, B, and C), 1 small cluster (D), and 1 singleton (E). Each cluster was defined to include MLVA26 types differing at <7 loci among the 26 loci. The 3 large clusters displayed distinct allelic diversity features. Eight loci (SS1, SS3, SS6, SS9, SS10, SS11, SS12, and SS23) had Simpson diversity values >0.5 for >1 of the 3 large clusters (Table). Differences in diversity values >0.3 among the 3 clusters were observed for 9 of the 26 VNTRs. The largest difference was in 2 hypervariable VNTRs (SS1 and SS6). These 2 VNTRs displayed a high degree of allelic diversity in cluster A, but were invariant in cluster B. SS1 was invariant but SS6 displayed a high degree of diversity in cluster C.
Of 1,672 isolates, 66% (1,100) were obtained from patients who acquired infections in Taiwan. Most isolates belonged to an insertion element IS1 interspacer 1 clone (6,7) that had slightly lower diversity values for some VNTRs than diversity values for total isolates obtained from a panel of 620 isolates representing the 620 MLVA26 types. However, a large number of clonal isolates from Taiwan did not affect relative magnitudes of diversity of the 26 VNTRs.
Although highly variable VNTRs are useful markers in distinguishing closely related strains, they are less useful for investigating clonal relationships among strains that have evolved over time (6). MLVA18 profiles, which excluded the data of 8 highly variable VNTRs (SS1, SS3, SS6, SS9, SS10, SS11, SS12, and SS23) from the 26-locus panel, were used to investigate the clonal structure of the isolates. On the basis of 18-locus profiles, 105 MLVA18 types were identified.
Figure
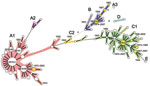
Figure. Clonal structure of 200 Shigella sonnei isolates. These isolates, representative of the 1,672 isolates analyzed in this study, were selected by obtaining 1 isolate for 1 multilocus variable number tandem repeat...
A simplified MST was created by analysis of a subset of 200 isolates selected by obtaining 1 MLVA18 type among those identified in each of 50 countries. As shown in the MST (Figure), cluster A was further divided into subclusters A1 and A2 and singleton A3, and cluster C was divided into subclusters C1 and C2.
Cluster A consisted of 46 MLVA18 types, which represented 1,382 isolates obtained since 1943 in 40 countries on 5 continents and the Pacific region (Table A1). Several MLVA18 types within cluster A were widespread. SS18.2 was detected in 23 countries on 5 continents. SS18.2 had 14 single-locus variants (SLVs) at which genotypes differed only at 1 of the 18 loci; 3 (SS18.80, SS18.1, and SS18.60) of the SLVs were detected in 13, 10, and 7 countries, respectively. SS18.2 and its SLVs represented 1,290 isolates obtained in during 1995–2008 from 38 countries on 5 continents. Four MLVA18 types were detected in samples obtained in 1943–1983. These isolates shared identical MLVA18 profiles or differed at 1–2 loci from recently obtained isolates. Subcluster A2 consisted of 4 MLVA18 types found in isolates from Argentina only. Singleton A3, which was found in isolates obtained in Vietnam in 2008, was distantly separate from subclusters A1 and A2.
Cluster B consisted of 12 MLVA18 types representing 75 isolates, which were obtained in 8 countries in Africa, Asia, and Europe. SS18.6 had the highest number of SLVs in cluster B and was detected in 5 countries in Asia and Africa. Five types were detected in isolates obtained in 1943–1974; they shared identical MLVA18 profiles or differed at 1–2 loci from recently recovered isolates.
Cluster C was relatively diverse; it consisted of 44 MLVA18 types, which represented 212 isolates obtained in 21 countries on 5 continents and the Pacific region. SS18.8 had the highest number of SLVs and was found in isolates obtained during 1974–2007 in 8 countries on 5 continents. SS18.4, the largest SLV of SS18.8, was found in isolates from 5 countries on 5 continents. Subcluster C2 consisted of isolates from Argentina obtained in 2002 and Sweden and Denmark in 1943. These isolates emerged in 1943–1974 and were genetically similar to recently obtained isolates. Clusters (clonal groups) A and C were globally spread, and clonal group B was found in countries in Africa, Asia and Europe only.
Cluster D contained 2 isolates obtained in French Guiana and Senegal in 2003. The isolate for singleton E was obtained in Malaysia in 1999.
Genetic analysis using MLVA presented a simple clonal structure for 1,672 S. sonnei isolates obtained since 1943 from 50 countries on 5 continents and the Pacific region. Three large clonal groups were identified; they displayed distinct allelic diversity features, particularly for 2 hypervariable VNTRs (SS1 and SS6). Clonal groups A and C were globally spread. One MLVA18 type (SS18.2) and several of its SLVs were widely distributed over 5 continents in the past 10 years.
Dr Filliol-Toutain is deputy director of the National Reference Center for Escherichia coli and Shigella at Institut Pasteur, Paris, France. Her research interests are surveillance of Shigella strains and Shiga toxin–producing E. coli strains circulating in France, detection of outbreaks, surveillance of the circulating serovars, and surveillance of antimicrobial drug resistance.
Acknowledgment
This study was supported by the Department of Health, Taiwan (grant no. DOH97-DC-2012).
References
- Gupta A, Polyak CS, Bishop RD, Sobel J, Mintz ED. Laboratory-confirmed shigellosis in the United States, 1989–2002: epidemiologic trends and patterns. Clin Infect Dis. 2004;38:1372–7. DOIPubMedGoogle Scholar
- Kotloff KL, Winickoff JP, Ivanoff B, Clemens JD, Swerdlow DL, Sansonetti PJ, Global burden of Shigella infections: implications for vaccine development and implementation of control strategies. Bull World Health Organ. 1999;77:651–66.PubMedGoogle Scholar
- Lewis HC, Kirk M, Ethelberg S, Stafford R, Olsen K, Nielsen EM, Outbreaks of shigellosis in Denmark and Australia associated with imported baby corn, August 2007–final summary. Euro Surveill. 2007;12:E071004 2.
- Ekdahl K, Andersson Y. The epidemiology of travel-associated shigellosis–regional risks, seasonality and serogroups. J Infect. 2005;51:222–9. DOIPubMedGoogle Scholar
- Liang SY, Watanabe H, Terajima J, Li CC, Liao JC, Tung SK, Multilocus variable-number tandem repeat analysis for molecular typing of Shigella sonnei. J Clin Microbiol. 2007;45:3574–80. DOIPubMedGoogle Scholar
- Chiou CS, Watanabe H, Wang YW, Wang WL, Terajima J, Thong KL, Utility of multilocus variable-number tandem-repeat analysis as a molecular tool for phylogenetic analysis of Shigella sonnei. J Clin Microbiol. 2009;47:1149–54. DOIPubMedGoogle Scholar
- Chiou CS, Wei HL, Wang YW, Liao JC, Li CC. Usefulness of inter-IS1 spacer polymorphisms for subtyping of Shigella sonnei isolates. J Clin Microbiol. 2006;44:3928–33. DOIPubMedGoogle Scholar
Figure
Tables
Cite This ArticleTable of Contents – Volume 17, Number 10—October 2011
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Chien-Shun Chiou, Centers for Disease Control, 5F, 20 Wen-Sin South Third Rd, Taichung 40855, Taiwan
Top