Autochthonous and Dormant Cryptococcus gattii Infections in Europe
Ferry Hagen

, M. Francisca Colom, Daniëlle Swinne, Kathrin Tintelnot, Roberta Iatta, Maria Teresa Montagna, Josep M. Torres-Rodriguez, Massimo Cogliati, Aristea Velegraki, Arjan Burggraaf, Alwin Kamermans, Johanna M. Sweere, Jacques F. Meis, Corné H.W. Klaassen, and Teun Boekhout
Author affiliations: Royal Netherlands Academy of Arts and Sciences Fungal Biodiversity Centre, Utrecht, the Netherlands (F. Hagen, A. Burggraaf, A. Kamermans, J.M. Sweere, T. Boekhout); University Medical Center Utrecht, Utrecht (F. Hagen, T Boekhout); Universidad Miguel Hernàndez, Alicante, Spain (M.F. Colom); Institute of Public Health, Brussels, Belgium (D. Swinne); Robert Koch-Institut, Berlin, Germany (K. Tintelnot); Università degli Studi di Bari, Bari, Italy (R. Iatta, M.T. Montagna); Autonomous University of Barcelona, Barcelona, Spain (J.M. Torres-Rodriguez); Università degli Studi di Milano, Milan, Italy (M. Cogliati); University of Athens, Athens, Greece (A. Velegraki); University College Utrecht, Utrecht (J.M. Sweere); Radboud University Nijmegen Medical Centre, Nijmegen, the Netherlands (J.F. Meis); and Canisius Wilhelmina Hospital, Nijmegen (J.F. Meis, C.H.W. Klaassen)
Main Article
Figure
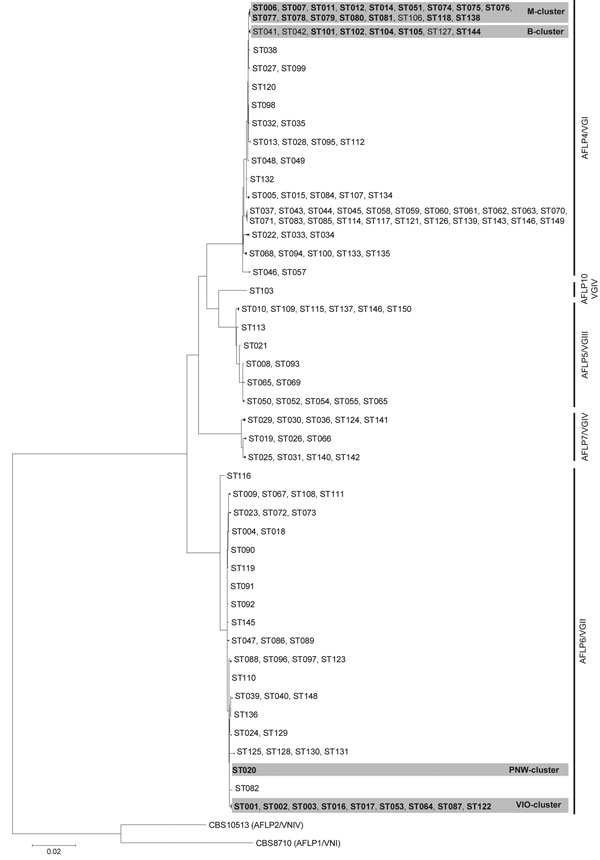
Figure. . . Maximum-likelihood phylogenetic analysis based on 10-loci multilocus sequence type data of Cryptococcus gattii isolates (condensed). Phylogenetic relatedness of 150 STs representing the 291 C. gattii isolates, calculated by using the maximum-likelihood algorithm and rooted by using the 2 C. neoformans reference strains CBS8710 (genotype AFLP1/VNI) and CBS10513 (genotype AFLP2/VNIV). Closely related sequence types were collapsed into 1 branch shown by multiple sequence type numbers. Boldface indicates sequence types that are within a shaded area belong to a specified C. gattii cluster; B, M, PNW, and VIO represent clusters from Brazil; Mediterranean Europe; the US Pacific Northwest outbreak; and the Vancouver Island, British Columbia, Canada, outbreak, respectively. AFLP, amplified fragment length polymorphism; ST, sequence type. Scale bar indicates number of substitutions per site. See for a detailed phylogenetic analysis.
Main Article
Page created: September 14, 2012
Page updated: September 14, 2012
Page reviewed: September 14, 2012
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.