Volume 18, Number 10—October 2012
Dispatch
Human Infection with Candidatus Neoehrlichia mikurensis, China
Figure 2
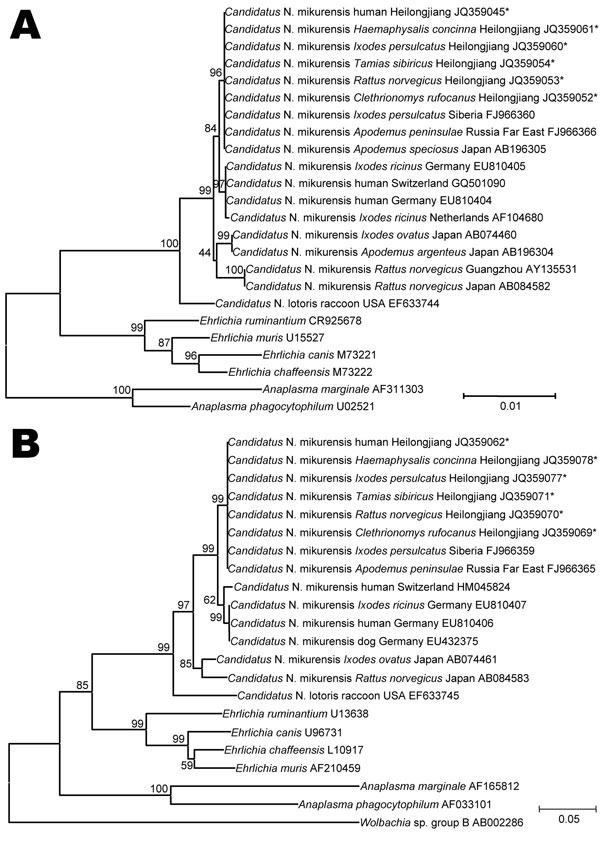
Figure 2. . . . A) Neighbor-joining trees based on the 16S rRNA gene (rrs) and B) the 60-kDa heat shock protein gene (groEL) of Candidatus Neoehrlichia mikurensis, China, generated by using Molecular Evolutionary Genetics Analysis software version 4.0, (www.megasoftware.net/) the maximum composite-likelihood method, and bootstrap analysis of 1,000 replicates. Asterisks indicate nucleotide sequences of Candidatus N. mikurensis determined in this study. Numbers on branches indicate percentage of replicates that reproduced the topology for each clade. Scale bars indicate estimated evolutionary distance. A total of 1,303 positions for rrs and 953 positions for groEL were analyzed. Sources of Candidatus N. mikurensis sequences are shown between species names and GenBank accession numbers.
1These authors contributed equally to this article.