Volume 18, Number 2—February 2012
Research
Characterization of Nipah Virus from Outbreaks in Bangladesh, 2008–2010
Michael K. Lo , Luis Lowe, Kimberly B. Hummel, Hossain M.S. Sazzad, Emily S. Gurley, M. Jahangir Hossain, Stephen P. Luby, David M. Miller, James A. Comer, Pierre E. Rollin, William J. Bellini, and Paul A. Rota
, Luis Lowe, Kimberly B. Hummel, Hossain M.S. Sazzad, Emily S. Gurley, M. Jahangir Hossain, Stephen P. Luby, David M. Miller, James A. Comer, Pierre E. Rollin, William J. Bellini, and Paul A. Rota
Figure 1
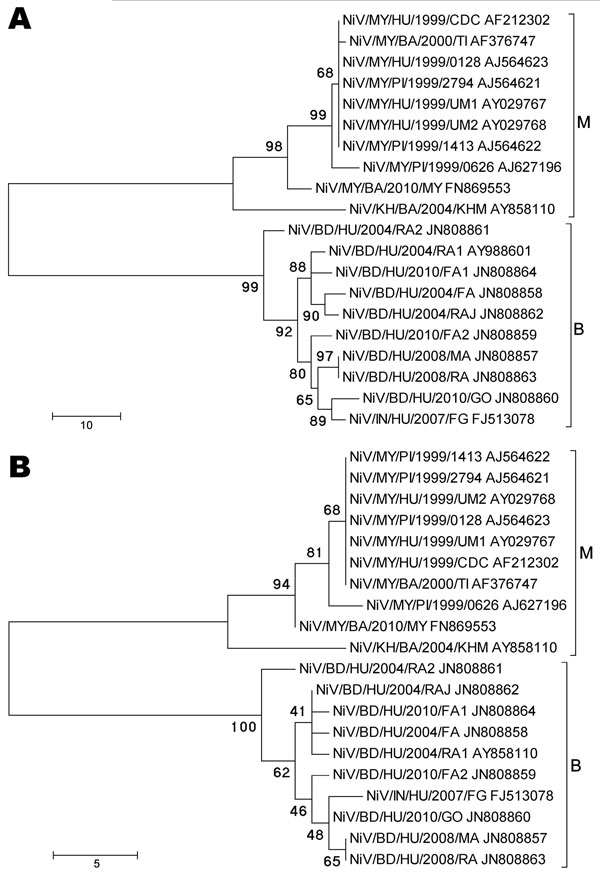
Figure 1. Phylogenetic analyses of sequences from the complete Nipah virus N ORF (A) and the 729-nt proposed N ORF genotyping window (B). Tree created with maximum parsimony, close-neighbor-interchange algorithm, 1,000 bootstrap replicates (16). Branch lengths are in units of number of changes over the whole sequence. Available GenBank accession numbers are shown for corresponding sequences. Proposed genotype groupings are indicated by brackets (M, B). ORF, open reading frame; MY, Malaysia; KH, Cambodia; BD, Bangladesh; IN, India; HU, human; PI, pig; BA, bat. Scale bars indicate number of sequence changes corresponding to illustrated branch length.
References
- Chua KB, Bellini WJ, Rota PA, Harcourt BH, Tamin A, Lam SK, Nipah virus: a recently emergent deadly paramyxovirus. Science. 2000;288:1432–5. DOIPubMedGoogle Scholar
- Chadha MS, Comer JA, Lowe L, Rota PA, Rollin PE, Bellini WJ, Nipah virus–associated encephalitis outbreak, Siliguri, India. Emerg Infect Dis. 2006;12:235–40.PubMedGoogle Scholar
- Arankalle VA, Bandyopadhyay BT, Ramdasi AY, Jadi R, Patil DR, Rahman M, Genomic characterization of Nipah virus, West Bengal, India. Emerg Infect Dis. 2011;17:907–9.PubMedGoogle Scholar
- Luby SP, Hossain MJ, Gurley ES, Ahmed B-N, Banu S, Khan SU, Recurrent zoonotic transmission of Nipah virus into humans, Bangladesh, 2001–2007. Emerg Infect Dis. 2009;15:1229–35. DOIPubMedGoogle Scholar
- International Centre for Diarrheal Disease Research. Bangladesh. Nipah outbreak in Faridpur District, Bangladesh, 2010. Health and Science Bulletin. 2010;8:6–11.
- Luby SP, Rahman M, Hossain MJ, Blum LS, Husain MM, Gurley E, Foodborne transmission of Nipah virus, Bangladesh. Emerg Infect Dis. 2006;12:1888–94. DOIPubMedGoogle Scholar
- Gurley ES, Montgomery JM, Hossain MJ, Bell M, Azad AK, Islam MR, Person-to-person transmission of Nipah virus in a Bangladeshi community. Emerg Infect Dis. 2007;13:1031–7.PubMedGoogle Scholar
- Luby SP, Gurley ES, Hossain MJ. Transmission of human infections with Nipah virus. Clin Infect Dis. 2009;49:1743–8. DOIPubMedGoogle Scholar
- Lo MK, Rota PA. The emergence of Nipah virus, a highly pathogenic paramyxovirus. J Clin Virol. 2008;43:396–400. DOIPubMedGoogle Scholar
- Harcourt BH, Lowe L, Tamin A, Liu X, Bankamp B, Bowden N, Genetic characterization of Nipah virus, Bangladesh, 2004. Emerg Infect Dis. 2005;11:1594–7.PubMedGoogle Scholar
- Rahman MA, Hossain MJ, Sultana S, Homaira N, Khan SU, Rahman M, Date palm sap linked to Nipah virus outbreak in Bangladesh, 2008. Vector Borne Zoonotic Dis. 2011; [Epub ahead of print]. DOIPubMedGoogle Scholar
- Daniels P, Ksiazek T, Eaton BT. Laboratory diagnosis of Nipah and Hendra virus infections. Microbes Infect. 2001;3:289–95. DOIPubMedGoogle Scholar
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–9. DOIPubMedGoogle Scholar
- Halpin K, Bankamp B, Harcourt BH, Bellini WJ, Rota PA. Nipah virus conforms to the rule of six in a minigenome replication assay. J Gen Virol. 2004;85:701–7. DOIPubMedGoogle Scholar
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;10:2731–9. DOIPubMedGoogle Scholar
- Ong ST, Yusoff K, Kho CL, Abdullah JO, Tan WS. Mutagenesis of the nucleocapsid protein of Nipah virus involved in capsid assembly. J Gen Virol. 2009;90:392–7. DOIPubMedGoogle Scholar
- Chan YP, Koh CL, Lam SK, Wang LF. Mapping of domains responsible for nucleocapsid protein-phosphoprotein interaction of Henipaviruses. J Gen Virol. 2004;85:1675–84. DOIPubMedGoogle Scholar
- Omi-Furutani M, Yoneda M, Fujita K, Ikeda F, Kai C. Novel phosphoprotein-interacting region in Nipah virus nucleocapsid protein and its involvement in viral replication. J Virol. 2010;84:9793–9. DOIPubMedGoogle Scholar
- Rodriguez JJ, Cruz CD, Horvath CM. Identification of the nuclear export signal and STAT-binding domains of the Nipah virus V protein reveals mechanisms underlying interferon evasion. J Virol. 2004;78:5358–67. DOIPubMedGoogle Scholar
- Shaw ML, Garcia-Sastre A, Palese P, Basler CF. Nipah virus V and W proteins have a common STAT1-binding domain yet inhibit STAT1 activation from the cytoplasmic and nuclear compartments, respectively. J Virol. 2004;78:5633–41. DOIPubMedGoogle Scholar
- Ciancanelli MJ, Volchkova VA, Shaw ML, Volchkov VE, Basler CF. Nipah virus sequesters inactive STAT1 in the nucleus via a P gene–encoded mechanism. J Virol. 2009;83:7828–41. DOIPubMedGoogle Scholar
- Rahman SA, Hassan SS, Olival KJ, Mohamed M, Chang LY, Hassan L, Characterization of Nipah virus from naturally infected Pteropus vampyrus bats, Malaysia. Emerg Infect Dis. 2010;16:1990–3.PubMedGoogle Scholar
- Ciancanelli MJ, Basler CF. Mutation of YMYL in the Nipah virus matrix protein abrogates budding and alters subcellular localization. J Virol. 2006;80:12070–8. DOIPubMedGoogle Scholar
- Patch JR, Han Z, McCarthy SE, Yan L, Wang LF, Harty RN, The YPLGVG sequence of the Nipah virus matrix protein is required for budding. Virol J. 2008;5:137. DOIPubMedGoogle Scholar
- Wang YE, Park A, Lake M, Pentecost M, Torres B, Yun TE, Ubiquitin-regulated nuclear-cytoplasmic trafficking of the Nipah virus matrix protein is important for viral budding. PLoS Pathog. 2010;6:e1001186. DOIPubMedGoogle Scholar
- Negrete OA, Chu D, Aguilar HC, Lee B. Single amino acid changes in the Nipah and Hendra virus attachment glycoproteins distinguish ephrinB2 from ephrinB3 usage. J Virol. 2007;81:10804–14. DOIPubMedGoogle Scholar
- Bowden TA, Aricescu AR, Gilbert RJ, Grimes JM, Jones EY, Stuart DI. Structural basis of Nipah and Hendra virus attachment to their cell-surface receptor ephrin-B2. Nat Struct Mol Biol. 2008;15:567–72. DOIPubMedGoogle Scholar
- Poch O, Blumberg BM, Bougueleret L, Tordo N. Sequence comparison of five polymerases (L proteins) of unsegmented negative-strand RNA viruses: theoretical assignment of functional domains. J Gen Virol. 1990;71:1153–62. DOIPubMedGoogle Scholar
- Harcourt BH, Tamin A, Halpin K, Ksiazek TG, Rollin PE, Bellini WJ, Molecular characterization of the polymerase gene and genomic termini of Nipah virus. Virology. 2001;287:192–201. DOIPubMedGoogle Scholar
- Rota PA, Featherstone DA, Bellini WJ. Molecular epidemiology of measles virus. Curr Top Microbiol Immunol. 2009;330:129–50. DOIPubMedGoogle Scholar
- Jin L, Rima B, Brown D, Orvell C, Tecle T, Afzal M, Proposal for genetic characterisation of wild-type mumps strains: preliminary standardisation of the nomenclature. Arch Virol. 2005;150:1903–9. DOIPubMedGoogle Scholar
- Wacharapluesadee S, Hemachudha T. Duplex nested RT-PCR for detection of Nipah virus RNA from urine specimens of bats. J Virol Methods. 2007;141:97–101. DOIPubMedGoogle Scholar
Page created: January 24, 2012
Page updated: January 24, 2012
Page reviewed: January 24, 2012
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.