Volume 19, Number 11—November 2013
Research
Atypical Scrapie Prions from Sheep and Lack of Disease in Transgenic Mice Overexpressing Human Prion Protein
Cite This Article
Citation for Media
Abstract
Public and animal health controls to limit human exposure to animal prions are focused on bovine spongiform encephalopathy (BSE), but other prion strains in ruminants may also have zoonotic potential. One example is atypical/Nor98 scrapie, which evaded statutory diagnostic methods worldwide until the early 2000s. To investigate whether sheep infected with scrapie prions could be another source of infection, we inoculated transgenic mice that overexpressed human prion protein with brain tissue from sheep with natural field cases of classical and atypical scrapie, sheep with experimental BSE, and cattle with BSE. We found that these mice were susceptible to BSE prions, but disease did not develop after prolonged postinoculation periods when mice were inoculated with classical or atypical scrapie prions. These data are consistent with the conclusion that prion disease is less likely to develop in humans after exposure to naturally occurring prions of sheep than after exposure to epizootic BSE prions of ruminants.
Bovine spongiform encephalopathy (BSE) is the transmissible spongiform encephalopathy (TSE) or prion disease of domestic cattle. The BSE prion is an epizootic agent and causes variant Creutzfeldt-Jakob disease (vCJD) in humans after dietary exposure (1–4). Because the time lag between exposure and development of vCJD may be decades, uncertainty about the extent of the pathogenicity of BSE for humans continues (5), and subclinical forms of infection may exist (6,7). A recent immunohistochemical study that estimated prevalence of prion infection in the UK population by screening samples from surgically removed appendixes found 1 in 2,000 persons were positive for the disease-associated form of the prion protein (PrP) (8). Similar uncertainty exists in our understanding of scrapie, the TSE of small ruminants, which has been heightened in recent years by finding BSE in goats (9,10), the possibility of BSE in sheep (11), and the discovery of atypical scrapie (12,13), a form of small-ruminant TSE, which had evaded statutory diagnosis until the early 2000s.
Recent analysis of surveillance data of TSEs in small ruminants in Great Britain, collected over the past 10 years, has demonstrated a dramatic decrease (up to 90%) in number of confirmed cases of classical scrapie in the national flock. However, atypical scrapie continues to affect sheep bred for their relative resistance to the classical form of this prion disease, and the proportion of sheep with resistant genotypes in the national flock is likely to have increased over the past decade because of the National Scrapie Plan for Great Britain. This increase has rekindled speculation that atypical scrapie in small ruminants might be a source of human prion disease (11). Although atypical scrapie has been discovered retrospectively in 2 UK sheep culled in 1987 and 1989 (14,15), the level and duration of human exposure to atypical scrapie prions are unknown, and this lack of knowledge confounds a cause-and-effect investigation of epidemiologic links between this animal disease and some form of CJD (11).
Over the past 2 decades, surrogate methods have been developed to assess the relative pathogenicity of animal prions for humans. One approach involves the experimental transmission of disease by inoculating homogenized brain tissue from affected animals into transgenic mice that are overexpressing 1 of the 2 common polymorphic forms of the human PrP (either methionine or valine at residue 129) on a mouse PrP null background (16). Such transgenic mice are fully susceptible to infection with human prions (16) and, to a lesser extent, cattle and ovine BSE prions (2,4,17), but appear resistant to chronic wasting disease prions from cervids (18–20). In this study, we inoculated transgenic mice that overexpressed human PrP with brain tissue from field sheep with natural cases of classical and atypical scrapie, sheep with serially-passaged experimental BSE, and cattle with BSE to assess the pathogenicity of natural scrapie prions relative to that of the known epizootic TSE agent, the cattle BSE prion strain.
Ovine and Bovine Prion Sources
From Great Britain’s Animal Health and Veterinary Laboratories Agency (AHVLA), we obtained 10% (w/v) brain homogenates prepared in sterile saline from sheep with neuropathologically confirmed prion disease and demonstrated ability to transmit prion disease to transgenic mice expressing ovine PrP or to wild-type mice (Table 1). We obtained scrapie-infected sheep brain from the Friedrich-Loeffler-Institut (Federal Research Institute for Animal Health, Germany) under a license granted by Department for Environment, Food and Rural Affairs, according to the terms of the Importation of Animal Pathogens Order 1980. Brain samples from sheep with neuropathologically confirmed cases of classical and atypical scrapie were prepared as 10% (w/v) homogenates in sterile Dulbecco phosphate-buffered saline lacking Ca2+ and Mg2+ ions (D-PBS) by extrusion through syringe needles of decreasing diameter. Brains from cattle with neuropathologically confirmed cases of BSE (collected specifically for transmission studies in the early 1990s) were provided by the UK Central Veterinary Laboratory (now AHVLA). We used 10% (w/v) homogenates prepared from the brainstems of 5 cattle with natural BSE to generate pooled inocula, designated I038, which was previously shown to transmit prion disease to wild-type FVB/N and C57Bl/6 mice, and to transgenic mice overexpressing human PrP (2,4,23,24). All experimental procedures involving ovine or bovine prions were carried out in a microbiological containment level 3 facility with strict adherence to safety protocols.
Transgenic Mice
Transgenic mice homozygous for a human PrP 129V transgene array and murine PrP null alleles (Prnpo/o), designated Tg(HuPrP129V+/+ Prnpo/o)-152 mice (129VV Tg152 mice), or homozygous for a human PrP 129M transgene array and murine PrP null alleles (Prnpo/o), designated Tg(HuPrP129M+/+ Prnpo/o)-35 mice (129MM Tg35 mice), have been described (1,2,4,24–26). Both lines of mice were used to generate FVB/N-HuPrP+/+ Prnpo/o congenic lines by backcrossing to FVB/N mice for 10 generations, followed by genetic testing (Charles River UK, Ltd., Margate, UK) by using 84 FVB-specific PCR microsatellite markers covering 19 chromosomes at ≈20-cM intervals, to select breeding pairs positive for 100% of the FVB-specific markers. Selected congenic pairs were interbred to remove the endogenous murine PrP gene and to establish homozygosity of the human PrP transgene array. The resulting congenic lines, designated 129MM Tg35c and 129VV Tg152c, overexpress human PrP in brain at levels of 2× and 6× that of pooled human brain, respectively.
Transmission Studies
Work with animals was performed under a license granted by the UK Home Office and conformed to institutional guidelines of the University College London and ARRIVE (Animal Research: Reporting In Vivo Experiments guidelines of The National Centre for the Replacement, Refinement and Reduction of Animals in Research). Brain homogenates (10% w/v) were diluted to 1% (w/v) in sterile D-PBS and passed through a 25-gauge needle. Each mouse was inoculated with 30-μL of 1% (w/v) brain homogenate because this avoids excessive animal losses within the first 48 hours postinoculation (4). Brain homogenates from prion-infected sheep were inoculated intracerebrally into groups of 20 transgenic mice that overexpressed human PrP. Thereafter, mice were examined daily and killed if they were exhibiting signs of distress or once a diagnosis of clinical prion disease was established (4,24,25). Clinical diagnosis can be confounded by nonspecific conditions that develop in mice as they age, and the mean lifespans of different lines of transgenic mice and the onset of aging artifacts vary greatly. On the basis of experience, we have limited these confounding effects by electively culling mice after postinoculation periods of >600 days. Notably, this also helps reduce the number of mice that die of old age, in which brain tissue can undergo autolytic deterioration that impairs immunohistochemical (IHC) analyses. At post-mortem, brains from inoculated mice were removed and divided sagittally, with half of the samples frozen and half fixed in formol-saline, and analyzed for abnormal PrP accumulation by IHC and immunoblotting.
Neuropathologic and Immunohistochemical Analyses
Brain fixed in 10% buffered formol-saline was immersed in 98% formic acid for 1 hour and embedded in paraffin wax. Serial sections (4-μm thick) were pretreated by boiling for 10 min in a low ionic strength buffer (2.1 mmol/L Tris, 1.3 mmol/L EDTA, 1.1 mmol/L sodium citrate, pH 7.8) before exposure to 98% formic acid for 5 min. Abnormal PrP accumulation was examined by using monoclonal antibody ICSM 35 against PrP (D-Gen Ltd., London, UK) on an automated IHC staining machine (Ventana Medical Systems, Inc., Tucson, AZ, USA) by using proprietary secondary detection reagents (Ventana Medical Systems, Inc.) before development with 3′3-diaminobenzedine tetrachloride as the chromogen (27). Conventional methods were used for Harris hematoxylin and eosin staining. Appropriate positive and negative controls were used throughout. Photographs were taken on an ImageView digital camera and composed with Adobe Photoshop (Adobe Systems, San Jose, CA, USA).
Immunoblotting
Proteinase K (PK) digestion (50 or 100 μg/mL final protease concentration, 1 hour, 37°C), electrophoresis, and immunoblotting of 10% (w/v) transgenic mouse brain homogenates or 10% (w/v) brain homogenates from sheep with classical scrapie (prepared in D-PBS) were performed as described (27,28). Human PrP or ovine PrP was detected by using monoclonal antibodies 3F4 (29) or ICSM 35 against PrP (D-Gen Ltd.), respectively. Mouse brain homogenates found negative for disease-related PrP (PrPSc) after analysis of 10 μL 10% (w/v) brain homogenate were reanalyzed by sodium phosphotungstic acid (NaPTA) precipitation of PrPSc (30) from 250 μL of 10% (w/v) brain homogenate as described (30).
Atypical scrapie sheep brain was analyzed by using the procedure of Gretzschel et al. (31,32) with modifications. In brief, 200 μL of 10% (w/v) brain homogenate in D-PBS was centrifuged at 500 × g for 5 min, after which the supernatant was discarded, and the pellet was resuspended to 100 μL final volume with D-PBS, followed by the addition of 100 μL 4% (w/v) sodium lauroylsarcosine (sarkosyl) in D-PBS. After incubation at 37°C for 30 min with constant agitation and centrifugation at 500 × g for 5 min, 150 μL of the supernatant was transferred to a new tube. The supernatant fraction was treated with 2 μL of Benzonase (Benzon nuclease purity 1; 25 U/μL; Merck, Nottingham, UK) for 30 min at 37°C with agitation and adjusted to a final concentration of 50 μg/mL PK (by adding 8 μL of a 1 mg/mL PK stock solution) and incubated at 37°C for 60 min with agitation. Samples were treated with 4 μL 100 mmol/L 4-(2-aminoethyl)-benzene sulfonyl fluoride, heated at 100°C for 5 min, adjusted with an equal volume of 2% (w/v) sarkosyl in D-PBS and 3 μL of Benzonase; they were then incubated for 30 min at 37°C with agitation before addition of 4% (w/v) NaPTA containing 170 mmol/L MgCl2, pH 7.4, to give a final concentration in the sample of 0.3% (w/v) NaPTA. After incubation for 60 min at 37°C, with constant agitation, samples were centrifuged at 16,100 × g for 30 min, and the supernatant fraction was discarded. The pellet fraction was resuspended to a final volume of 10 μL in D-PBS containing 0.1% (w/v) sarkosyl and analyzed by electrophoresis, immunoblotting, and high sensitivity chemiluminescence (27,28), using monoclonal antibody ICSM 35 against PrP to detect ovine PrP.
Scrapie Prions from Sheep and Lack of Disease in Transgenic Mice
Figure 1
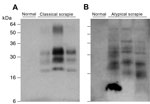
Figure 1. . Detection of disease-related prion protein (PrPSc) in brains of sheep with field cases of classical and atypical scrapie, Germany. Both panels show immunoblots of proteinase K–digested brain homogenate analyzed...
We examined classical and atypical scrapie sheep brain homogenates from UK field cases (AHVLA) that contain PK-resistant ovine PrPSc and efficiently transmitted clinical prion disease to transgenic mice expressing ovine PrP (21,22) (Table 1), together with a series of PK-resistant PrP-positive brain homogenates, from sheep in Germany with field cases of classical and atypical scrapie (Figure 1). All natural brain isolates examined produced no clinical prion disease or biochemical or histopathologic evidence for subclinical prion infection in transgenic mice that overexpressed human PrP after postinoculation intervals of >600 days (Table 2).
Consistent with the inability of IHC or high sensitivity immunoblotting to detect pathologic PrP in the brains of inoculated mice, neuropathologic examination of the brain showed no difference in spongiform change or gliosis from that observed in the brains of age-matched control mice (data not shown). From these findings, we conclude that both methionine and valine residue 129 variants of human PrP are refractory to pathologic conversion by these ovine prion strains in transgenic mice.
Transmission of Cattle BSE Prions to Transgenic Mice
Brain isolates from sheep with classical and atypical scrapie (including those with demonstrated prion infectivity in transgenic mice expressing ovine PrP) did not transmit prion disease to transgenic mice that were overexpressing human PrP. This fact contrasts markedly with the known susceptibility of these mice to transmission of multiple cattle BSE isolates (2,4,24,25) as well as to transmission of a wide range of human-acquired prion diseases (including kuru and vCJD) and sporadic prion disease isolates (2,4,24–26).
Figure 2
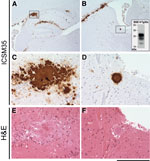
Figure 2. . Immunohistochemical analysis of cattle bovine spongiform encephalopathy (BSE) prion–infected 129MM Tg35c mouse brain. Hippocampal region (A) and striatum (B) from a transgenic 129MM Tg35c mouse with subclinical prion infection culled...
Concomitant with the current study, and as part of a separate experiment, we inoculated 129MM Tg35c mice intracerebrally with cattle BSE isolate I038. This BSE isolate has previously been shown to be transmissible to the parent 129MM Tg35 transgenic line, producing an attack rate of 8/20 inoculated mice (4) (Table 3). Affected 129MM Tg35 mice in these transmissions were culled (because of intercurrent illness or clinical prion disease) within 600 days of inoculation (Table 3) and demonstrated the presence of abnormal PrP in brain by IHC and immunoblotting (4). In 129MM Tg35c mice, cattle BSE isolate I038 produced an attack rate of 5/12 in intracerebrally inoculated mice (Table 3). Infection was characterized by the detection of abnormal PrP by IHC (Figure 2, panels A, B), which included large amorphous PrP deposits (Figure 2, panels C, E) and florid PrP plaques (Figure 2, panels D, F), and the detection of type 4 PrPSc in brain homogenate by immunoblotting (Figure 2, panel B inset). Intercurrent illness before 600 days postinoculation was seen in only one 129MM Tg35c mouse, with the remaining mice in the group (11/12) culled 611–853 days postinoculation (Table 3). Although most mice survived >600 days after inoculation, the attack rate of cattle BSE isolate I038 in 129MM Tg35c mice remained the same as observed in the parental 129MM Tg35 mouse line with ≈40% of inoculated mice becoming infected (Table 3). In addition, we found that 129MM Tg35 and 129MM Tg35c mice showed equivalent susceptibilities (100% attack rates) to vCJD or classical CJD prions (Table 3).
Experimental Ovine BSE in Transgenic Mice Expressing Human PrP 129 Methionine
Figure 3
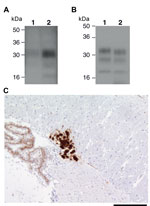
Figure 3. . Ovine bovine spongiform encephalopathy (BSE) prion transmission to a 129MM Tg35c mouse. Panel A shows immunoblot detection of disease-related prion protein (PrPSc) in 10 μL of proteinase K (PK)–digested 10%...
Recently, 2 studies have concluded that experimental sheep BSE prions may propagate more efficiently than cattle BSE prions in transgenic mice that express human PrP 129 methionine (17,34). One of these studies convincingly established that sheep and goat BSE prions transmitted a molecular and neuropathologic phenotype congruent with transmission of vCJD prions in the same mice (17). These data strongly suggest that small ruminant BSE prions could act as causal agents of vCJD (17). In this study, we also examined the transmission properties of 2 experimental sheep BSE brain isolates derived from the primary transmission and secondary passage of cattle BSE in sheep. These AHVLA isolates were provided as brain homogenates that contained PK-resistant ovine PrP (Figure 3, panel A) and had known ability to transmit clinical prion disease to wild-type RIII mice (Table 1).
In the transgenic mice expressing human PrP, clinical prion disease was not produced by either of the 2 experimental sheep BSE isolates after postinoculation intervals >600 days (Table 2). Examination of brain from these inoculated mice by IHC and immunoblotting, after NaPTA precipitation of brain homogenate, showed that only a single 129MM Tg35c recipient of the secondary passage ovine BSE isolate had evidence of subclinical prion infection (Tables 1, 2; Figure 3). This mouse was culled 661 days postinoculation when the experiment was terminated. PrPSc was detectable in the brain of this transgenic mouse without requirement for NaPTA precipitation for detection and appeared similar (but not identical) to type 4 PrPSc seen in vCJD brain (Figure 3, panel B). Florid PrP plaques were not observed, and abundant PrP deposits were restricted to the corpus callosum (Figure 3, panel C), accompanied by occasional punctate PrP deposits in the cortex and sparse diffuse PrP deposits in the thalamus and hypothalamus (data not shown). Secondary passages of this isolate in additional human PrP–expressing transgenic mice and wild-type FVB/N mice have been initiated to comprehensively define prion strain type.
Why the efficiency of transmission of experimental sheep BSE prions to 129MM Tg35c mice is low compared with that reported in different lines of human PrP 129 methionine–expressing mice (17,34) is unclear. One possible reason may simply relate to the prion titers in the inocula. Plinston et al. reported that 2 different inocula prepared from the same experimental sheep BSE brain had markedly different transmission efficiencies to gene-targeted mice expressing human PrP 129 methionine at endogenous levels (34). However, all AHVLA ovine prion isolates used in this study were chosen because they produced short survival periods and high attack rates in either ovine PrP transgenic mice or wild type mice (Table 1). Therefore, other possibilities must also be considered. In particular, studies involving different laboratories use different lines of genetically modified mice. Variation in genetic background and differences in PrP expression levels are known to influence host susceptibility to prion infection (16).
In this study, we have shown that disease does not develop in transgenic mice overexpressing human PrP when mice are inoculated with ovine prions from sheep with natural cases of classical scrapie and atypical scrapie from Great Britain and Germany. These transgenic mice are susceptible to infection, and clinical disease develops when mice are challenged with brain tissue from cattle affected by classical BSE (2,4,24,25) or brain tissue from humans affected by classical (sporadic and iatrogenic) CJD, kuru, or vCJD (2,4,24–26). Therefore, this suggests that the transmission barrier associated with the interaction of human PrP and the prion strain causing epizootic BSE in cattle is lower than that associated with the prion strain causing atypical scrapie in sheep. Serial, blind passage of brain homogenates from “negative” challenged mice from this experiment into other lines of transgenic mice expressing either human PrP or ovine PrP will now be required to determine whether this transmission barrier is absolute.
Our findings complement those of other recent studies that have investigated the zoonotic potential of ruminant prion strains using other lines of human PrP–expressing mice. Gene-targeted human PrP–expressing mice have been shown to be resistant to infection with classical and atypical scrapie prions from sheep (34,35) and BSE prions from cattle (36) but are susceptible to infection with BSE prions from sheep (34). Transgenic mice with 6-fold overexpression of human PrP 129 methionine are susceptible to infection with cattle BSE prions but show greater susceptibility to ovine and caprine BSE prions (17).
Although we found evidence for transmission of experimental ovine BSE to transgenic mice expressing human PrP 129 methionine, the relative attack rate was lower than observed in the other lines of mice (17,34). The reasons underlying this are not clear but may relate to differences in the prion isolates themselves or differences in the various lines of mice. To definitively investigate interlaboratory differences in the apparent behavior of ovine BSE prions and reach a consensus, a panel of ovine prion inocula would need to formally undergo endpoint titration across the different lines of humanized mice and also in ovine PrP–expressing transgenic mice.
No strain variation has been found so far in the transmission, biochemical, or histopathologic characteristics of atypical scrapie prions (22,37), and so inferences from the present study are not confounded by sampling or strain considerations. This is not so for cases of classical scrapie and, although our findings on atypical scrapie prions indicate that the zoonotic potential of this ovine prion strain is lower than for ruminant BSE prions, further transmission studies using a wider variety of field cases of classical scrapie are required to provide further reassurance of the low or negligible zoonotic potential of all sheep prions. Examining extraneural tissues (in particular, the spleen) in ovine prion-challenged mice will be critical because recent findings have shown that cross-species prion transmission efficacy can exhibit a dramatic tissue-dependence in the same host (38).
Dr Wadsworth is a program leader within the UK Medical Research Council Prion Unit in London. His primary research interest is the molecular basis of mammalian prion strains.
Acknowledgments
We thank all patients and their families for generously consenting to the use of human tissues in this research. We also thank our biological services team for animal care and R. Young for preparing the figures.
Some of the work was undertaken at University College London Hospital National Health Service Foundation Trust, which received a proportion of funding from the Department of Health’s National Institute for Health Research Biomedical Research Centre. This research was funded by the Medical Research Council (UK) and the European Union.
J.C. is a director and J.C. and J.D.F.W. are shareholders and consultants of D-Gen Limited, an academic spin-out company working in the field of prion disease diagnosis, decontamination, and therapeutics. D-Gen markets the ICSM35 antibody used in this study.
References
- Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature. 1996;383:685–90. DOIPubMedGoogle Scholar
- Hill AF, Desbruslais M, Joiner S, Sidle KCL, Gowland I, Collinge J. The same prion strain causes vCJD and BSE. Nature. 1997;389:448–50. DOIPubMedGoogle Scholar
- Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature. 1997;389:498–501. DOIPubMedGoogle Scholar
- Asante EA, Linehan J, Desbruslais M, Joiner S, Gowland I, Wood A, BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J. 2002;21:6358–66. DOIPubMedGoogle Scholar
- Collinge J, Whitfield J, McKintosh E, Beck J, Mead S, Thomas DJ, Kuru in the 21st century—an acquired human prion disease with very long incubation periods. Lancet. 2006;367:2068–74. DOIPubMedGoogle Scholar
- Hilton DA, Ghani AC, Conyers L, Edwards P, McCardle L, Ritchie D, Prevalence of lymphoreticular prion protein accumulation in UK tissue samples. J Pathol. 2004;203:733–9. DOIPubMedGoogle Scholar
- Clewley JP, Kelly CM, Andrews N, Vogliqi K, Mallinson G, Kaisar M, Prevalence of disease related prion protein in anonymous tonsil specimens in Britain: cross sectional opportunistic survey. BMJ. 2009;338:b1442. DOIPubMedGoogle Scholar
- UK Health Protection Agency. Summary results of the second national survey of abnormal prion prevalence in archived appendix specimens. Heath Protection Agency Report. News Archives. 2012;6:32 [cited 2012 Aug 10]. http://www.hpa.org.uk/hpr/archives/2012/news3212.htm#bnrmlp
- Eloit M, Adjou K, Coulpier M, Fontaine JJ, Hamel R, Lilin T, BSE agent signatures in a goat. Vet Rec. 2005;156:523–4 .PubMedGoogle Scholar
- Spiropoulos J, Lockey R, Sallis RE, Terry LA, Thorne L, Holder TM, Isolation of prion with BSE properties from farmed goat. Emerg Infect Dis. 2011;17:2253–61. DOIPubMedGoogle Scholar
- European Food Safety AuthorityPanel on Biological Hazards (BIOHAZ). Joint scientific opinion on any possible epidemiological or molecular association between TSEs in animals and humans. EFSA Journal. 2011;9:1945–2056 [cited 11 Sep 2013]. http://www.efsa.europa.eu/en/publications/efsajournal.htm
- Benestad SL, Sarradin P, Thu B, Schonheit J, Tranulis MA, Bratberg B. Cases of scrapie with unusual features in Norway and designation of a new type, Nor98. Vet Rec. 2003;153:202–8. DOIPubMedGoogle Scholar
- Benestad SL, Arsac JN, Goldmann W, Noremark M. Atypical/Nor98 scrapie: properties of the agent, genetics, and epidemiology. Vet Res. 2008;39:19. DOIPubMedGoogle Scholar
- Webb PR, Powell L, Denyer M, Marsh S, Weaver C, Simmons MM, A retrospective immunohistochemical study reveals atypical scrapie has existed in the United Kingdom since at least 1987. J Vet Diagn Invest. 2009;21:826–9. DOIPubMedGoogle Scholar
- Bruce ME, Nonno R, Foster J, Goldmann W, Di Bari M, Esposito E, Nor98-like sheep scrapie in the United Kingdom in 1989. Vet Rec. 2007;160:665–6. DOIPubMedGoogle Scholar
- Wadsworth JD, Asante EA, Collinge J. Contribution of transgenic models to understanding human prion disease. Neuropathol Appl Neurobiol. 2010;36:576–97. DOIPubMedGoogle Scholar
- Padilla D, Beringue V, Espinosa JC, Andreoletti O, Jaumain E, Reine F, Sheep and goat BSE propagate more efficiently than cattle BSE in human PrP transgenic mice. PLoS Pathog. 2011;7:e1001319. DOIPubMedGoogle Scholar
- Kong Q, Huang S, Zou W, Vanegas D, Wang M, Wu D, Chronic wasting disease of elk: transmissibility to humans examined by transgenic mouse models. J Neurosci. 2005;25:7944–9. DOIPubMedGoogle Scholar
- Tamgüney G, Giles K, Bouzamondo-Bernstein E, Bosque PJ, Miller MW, Safar J, Transmission of elk and deer prions to transgenic mice. J Virol. 2006;80:9104–14. DOIPubMedGoogle Scholar
- Sandberg MK, Al Doujaily H, Sigurdson CJ, Glatzel M, O’Malley C, Powell C, Chronic wasting disease prions are not transmissible to transgenic mice overexpressing human prion protein. J Gen Virol. 2010;91:2651–7. DOIPubMedGoogle Scholar
- Thackray AM, Hopkins L, Spiropoulos J, Bujdoso R. Molecular and transmission characteristics of primary passaged ovine scrapie isolates in conventional and ovine PrP transgenic mice. J Virol. 2008;82:11197–207. DOIPubMedGoogle Scholar
- Griffiths PC, Spiropoulos J, Lockey R, Tout AC, Jayasena D, Plater JM, Characterisation of atypical scrapie cases from Great Britain in transgenic ovine PrP mice. J Gen Virol. 2010;91:2132–8. DOIPubMedGoogle Scholar
- Lloyd SE, Linehan J, Desbruslais M, Joiner S, Buckell J, Brandner S, Characterization of two distinct prion strains derived from bovine spongiform encephalopathy transmissions to inbred mice. J Gen Virol. 2004;85:2471–8. DOIPubMedGoogle Scholar
- Wadsworth JD, Asante EA, Desbruslais M, Linehan J, Joiner S, Gowland I, Human prion protein with valine 129 prevents expression of variant CJD phenotype. Science. 2004;306:1793–6. DOIPubMedGoogle Scholar
- Asante EA, Linehan J, Gowland I, Joiner S, Fox K, Cooper S, Dissociation of pathological and molecular phenotype of variant Creutzfeldt-Jakob disease in transgenic human prion protein 129 heterozygous mice. Proc Natl Acad Sci U S A. 2006;103:10759–64. DOIPubMedGoogle Scholar
- Wadsworth JD, Joiner S, Linehan JM, Desbruslais M, Fox K, Cooper S, Kuru prions and sporadic Creutzfeldt-Jakob disease prions have equivalent transmission properties in transgenic and wild-type mice. Proc Natl Acad Sci U S A. 2008;105:3885–90. DOIPubMedGoogle Scholar
- Wadsworth JD, Powell C, Beck JA, Joiner S, Linehan JM, Brandner S, Molecular diagnosis of human prion disease. Methods Mol Biol. 2008;459:197–227. DOIPubMedGoogle Scholar
- Wadsworth JD, Joiner S, Hill AF, Campbell TA, Desbruslais M, Luthert PJ, Tissue distribution of protease resistant prion protein in variant CJD using a highly sensitive immuno-blotting assay. Lancet. 2001;358:171–80. DOIPubMedGoogle Scholar
- Kascsak RJ, Rubenstein R, Merz PA, Tonna DeMasi M, Fersko R, Carp RI, Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol. 1987;61:3688–93 .PubMedGoogle Scholar
- Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Eight prion strains have PrPSc molecules with different conformations. Nat Med. 1998;4:1157–65. DOIPubMedGoogle Scholar
- Gretzschel A, Buschmann A, Eiden M, Ziegler U, Luhken G, Erhardt G, Strain typing of German transmissible spongiform encephalopathies field cases in small ruminants by biochemical methods. J Vet Med B Infect Dis Vet Public Health. 2005;52:55–63. DOIPubMedGoogle Scholar
- Gretzschel A, Buschmann A, Langeveld J, Groschup MH. Immunological characterization of abnormal prion protein from atypical scrapie cases in sheep using a panel of monoclonal antibodies. J Gen Virol. 2006;87:3715–22. DOIPubMedGoogle Scholar
- Hill AF, Joiner S, Wadsworth JD, Sidle KC, Bell JE, Budka H, Molecular classification of sporadic Creutzfeldt-Jakob disease. Brain. 2003;126:1333–46. DOIPubMedGoogle Scholar
- Plinston C, Hart P, Chong A, Hunter N, Foster J, Piccardo P, Increased susceptibility of human-PrP transgenic mice to bovine spongiform encephalopathy following passage in sheep. J Virol. 2011;85:1174–81 . DOIPubMedGoogle Scholar
- Wilson R, Plinston C, Hunter N, Casalone C, Corona C, Tagliavini F, Chronic wasting disease and atypical forms of BSE and scrapie are not transmissible to mice expressing wild-type levels of human PrP. J Gen Virol. 2012;93:1624–9 . DOIPubMedGoogle Scholar
- Bishop MT, Hart P, Aitchison L, Baybutt HN, Plinston C, Thomson V, Predicting susceptibility and incubation time of human-to-human transmission of vCJD. Lancet Neurol. 2006;5:393–8 . DOIPubMedGoogle Scholar
- Götte DR, Benestad SL, Laude H, Zurbriggen A, Oevermann A, Seuberlich T. Atypical scrapie isolates involve a uniform prion species with a complex molecular signature. PLoS ONE. 2011;6:e27510 . DOIPubMedGoogle Scholar
- Béringue V, Herzog L, Jaumain E, Reine F, Sibille P, Le Dur A, Facilitated cross-species transmission of prions in extraneural tissue. Science. 2012;335:472–5 . DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 19, Number 11—November 2013
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Jonathan D.F. Wadsworth, Institute of Neurology, University College London, Medical Research Council Prion Unit, Queen Square, London WC1N 3BG, United Kingdom
Top