Volume 19, Number 8—August 2013
Dispatch
Whole Genome Sequencing of an Unusual Serotype of Shiga Toxin–producing Escherichia coli
Cite This Article
Citation for Media
Abstract
Shiga toxin–producing Escherichia coli serotype O117:K1:H7 is a cause of persistent diarrhea in travelers to tropical locations. Whole genome sequencing identified genetic mechanisms involved in the pathoadaptive phenotype. Sequencing also identified toxin and putative adherence genes flanked by sequences indicating horizontal gene transfer from Shigella dysenteriae and Salmonella spp., respectively.
There are >400 serotypes of Shiga toxin–producing Escherichia coli (STEC), and >100 of these are known to be associated with severe disease in humans (1). STEC are defined by the presence of 1 or both phage-encoded Shiga toxin genes stx1 and stx2. However, those serotypes associated with more severe disease generally harbor additional virulence genes, such as eae (intimin), which is encoded on the locus of enterocyte effacement, or virulence regulation genes, such as aggR, which is located on the aggregative adherence plasmid. Both of these genes mediate attachment of the bacteria to the host gut mucosa (2). The stx1 gene is also found in Shigella dysenteriae serotype 1.
A range of molecular typing methods show that the shigellae belong within the Escherichia coli species (3). Peng et al. (4) described an evolutionary path of Shigella spp. from E. coli involving gene acquisition (virulence plasmid and pathogenicity islands) and gene loss (pathoadaptivity). Gene loss, or loss of gene function, may result from changes to bacterial biosynthesis pathways driven by the abundance of resources in the host or because the genes may encode proteins adverse to bacterial virulence.
Olesen et al. (5) described a strain of STEC serotype O117:K1:H7 found in travelers from Denmark who returned from tropical locations. The strain was unusual because it was negative for the production of lysine decarboxylase and β-galactosidase (ortho-nitrophenol test) and positive only for stx1.
Since 2004, 19 isolates of STEC O117:K1:H7 have been submitted to the Gastrointestinal Bacteria Reference Unit at the Health Protection Agency in London, UK, from frontline diagnostic microbiology laboratories in England and Wales for confirmation of identification and typing (Table). All isolates were originally misidentified by the submitting laboratory as Shigella sonnei or Shigella spp., probably because of the unusual biochemical phenotype exhibited by this strain. The purpose of this study was to use whole genome sequencing to investigate the evolutionary origins, putative virulence genes, and pathoadaptive mechanisms of this unusual STEC serotype.
DNA from 5 isolates (151/06, 371/08, 290/10, 754/10, and 229/11) was prepared for sequencing by using the Nextera sample preparation method and sequenced with a standard 2 × 151 base protocol on a MiSeq instrument (Illumina, San Diego, CA, USA) (6). Sequences were analyzed as described (7). In brief, Velvet version 1.1.04 (www.ebi.ac.uk/~zerbino/velvet/) was used to produce an average of 489 contigs with an average N50 length of 38722. Illumina reads were mapped to the reference strain (GenBank accession no. CU928145) by using Bowtie2 2.0.0 β-5 (http://bowtie-bio.sourceforge.net/bowtie2/) and a variant call format file was created from each of the binary alignment maps, which were further parsed to extract only single nucleotide polymorphism (SNP) positions that were of high quality in all genomes.
Figure
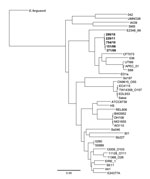
Figure. . . Maximum-likelihood dendrogram for 5 strains of Shiga toxin–producing Escherichia coli serotype O117 in the Gastrointestinal Bacteria Reference Unit (Health Protection Agency, London, UK) archive (boldface), 32 other E. coli...
Concatenated SNPs generated against the reference strain 55989 were used to produce a maximum-likelihood phylogeny of 5 strains in the Gastrointestinal Bacteria Reference Unit archive and 36 other publically available E. coli genomes and Shigella spp. (Figure). Despite temporal and spatial diversity of the 5 sequenced isolates, they clustered on the same branch, but they were distant from other publically available sequences of STEC strains.
A phylogenetic tree based on a diverse range of E. coli showed that the 5 strains of STEC O117 have 130 polymorphic positions, and the closest 2 strains (299/11 and 754/10) are 26 SNPs apart (Table; Figure). Furthermore, on the basis of a diverse range of E. coli, genome sequences of EDL933 and Sakai, 2 well-described strains of STEC O157, are ≈35 SNPs apart. The multilocus sequence type ST504 was assigned in accordance with the E. coli multilocus sequence type databases at the Environment Research Institute, University College (Cork, Ireland).
Alignment of the genome of strain 229/11 with STEC O157 (EDL933) and Shigella dystenteriae serotype 1 (Sd197) indicated gene acquisition, loss, and rearrangement in 229/11. The stx1 gene is adjacent to the yjhS gene in 229/11 and Sd197, and in 229/11 this fragment is flanked by phage-like sequences that are closely related to Stx2-converting phage sequences but not to other Stx1-converting phages. This unusual gene arrangement was described by Sato et al. (8). In Sd197, this region is flanked by integrases and insertion sequences. Other open reading frames homologous to those of Shigella spp. in stx-flanking regions in E. coli have been described, and it is likely that E. coli and the shigellae have exchanged stx many times in their evolutionary past but only certain strains, such as 229/11, have the appropriate genomic background to retain and stably express Stx (9).
Strain 229/11 also contains a 10-kb pathogenicity island (PAI) harboring the ratA, Sivl, and SivH genes and shares homology with PAI CS54 found in Salmonella spp. (10) and a PAI found in avian pathogenic E. coli (11). SivH has been described as similar to the intimin gene (10). SivH may facilitate attachment to the host gut mucosa and could explain the long persistence of STEC O117:K1:H7 in infected patients (5). In vitro inactivation of sivH in S. enterica serovar Typhimurium resulted in a reduced ability to colonize Peyer’s patches (10). In S. enterica serovar Typhimurium, CS54 is 25-kb and encodes shdA, ratA, ratB, sivl, and sivH, whereas in S. enterica subsp. II, S. bongori serotypes and 229/11, ratB, and shdA are absent (10).
Cadaverine has an inhibitory effect on enterotoxin activity by preventing full expression of the virulent phenotype, and it has been suggested that there is evolutionary pressure to mutate or delete the cadA gene (12). This gene is missing from S. flexneri (Sf301) and S. boydii (Sb227) because of inversion-associated deletions, and in Sd197 and S. sonnei (Ss046) it is inactivated by a frameshift mutation and an insertion sequence, respectively (12). In 229/11, loss of cadA (lysine decarboxylase) activity is caused by repositioning of the of the cadA activator gene, CadC, upstream of the cadA gene and a 90-bp deletion at the 5′ end of cadC. The cadA gene and truncated cadC gene are separated by a large fragment of DNA inserted into the cadC gene. This fragment contains several open reading frames, including genes encoding aerobactin siderophore biosynthesis proteins.
Lactose fermentation is a biochemical property commonly used for distinguishing Shigella spp. from E. coli because shigellae are non- or late-lactose fermenters. In Sd197 and Ss046 (late lactose–fermenting strains), the key gene, lacZ (encoding β-
E. coli as a species contains a large diversity of adaptive paths. This diversity is the result of a highly dynamic genome, with a constant and frequent flux of insertions and deletions (3). Pathogenicity in STEC O117:K1:H7 is most likely multifactorial and results from a novel combination of lack of cadA and lacZ expression and the presence of stx1 and the intimin-like sivH genes, demonstrating pathoadaptivity and horizontal gene transfer.
Dr. Dallman is lead bioinformatician in the Gastrointestinal Bacterial Reference Unit at the Health Protection Agency in London, UK. His primary research interest is application of whole genome sequencing of enteric pathogens to aid public health investigations.
Acknowledgments
We thank the Health Protection Agency Next-Generation Sequencing Implementation Group for support and Flemming Scheutz for helpful discussions.
This study was supported by the Health Protection Agency Strategic Research and Development Fund (grant no. 108061).
References
- Bergey’s manual of systematic bacteriology. The Proteobacteria, 2nd ed. Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. New York: Springer; 2005.
- Kaper JB, Nataro JP, Mobley HL. Pathogenic Escherichia coli. Nat Rev Microbiol. 2004;2:123–40 and. DOIPubMedGoogle Scholar
- Kaas RS, Friis C, Ussery DW, Aarestrup FM. Estimating variation within the genes and inferring the phylogeny of 186 sequenced diverse Escherichia coli genomes. BMC Genomics. 2012;13:577 and. DOIPubMedGoogle Scholar
- Peng J, Yang J, Jin Q. The molecular evolutionary history of Shigella spp. and enteroinvasive Escherichia coli. Infect Genet Evol. 2009;9:147–52 and. DOIPubMedGoogle Scholar
- Olesen B, Jensen C, Olsen K, Fussing V, Gerner-Smidt P, Scheutz F. VTEC O117:K1:H7. A new clonal group of E. coli associated with persistent diarrhoea in Danish travellers. Scand J Infect Dis. 2005;37:288–94 and. DOIPubMedGoogle Scholar
- Köser CU, Holden MT, Ellington MJ, Cartwright EJ, Brown NM, Ogilvy-Stuart AL, Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N Engl J Med. 2012;366:2267–75 and. DOIPubMedGoogle Scholar
- Dallman T, Smith GP, O'Brien B, Chattaway MA, Finlay D, Grant KA, Characterization of a verocytotoxin-producing enteroaggregative Escherichia coli serogroup O111:H21 strain associated with a household outbreak in Northern Ireland. J Clin Microbiol. 2012;50:4116–9 and. DOIPubMedGoogle Scholar
- Sato T, Shimizu T, Watarai M, Kobayashi M, Kano S, Hamabata T, Genome analysis of a novel Shiga toxin 1 (Stx1)–converting phage which is closely related to Stx2-converting phages but not to other Stx1-converting phages. J Bacteriol. 2003;185:3966–71 and. DOIPubMedGoogle Scholar
- Escobar-Páramo P, Clermont O, Blanc-Potard AB, Bui H, Le Bouguénec C, Denamur E. A specific genetic background is required for acquisition and expression of virulence factors in Escherichia coli. Mol Biol Evol. 2004;21:1085–94 and. DOIPubMedGoogle Scholar
- Kingsley RA, Humphries AD, Weening EH, De Zoete MR, Winter S, Papaconstantinopoulou A, Molecular and phenotypic analysis of the CS54 island of Salmonella enterica serotype Typhimurium: identification of intestinal colonization and persistence determinants. Infect Immun. 2003;71:629–40 and. DOIPubMedGoogle Scholar
- Schouler C, Koffmann F, Amory C, Leroy-Sétrin S, Moulin-Schouleur M. Genomic subtraction for the identification of putative new virulence factors of an avian pathogenic Escherichia coli strain of O2 serogroup. Microbiology. 2004;150:2973–84 and. DOIPubMedGoogle Scholar
- Yang F, Yang J, Zhang X, Chen L, Jiang Y, Yan Y, Genome dynamics and diversity of Shigella species, the etiologic agents of bacillary dysentery. Nucleic Acids Res. 2005;33:6445–58 and. DOIPubMedGoogle Scholar
- Bliven KA, Maurelli AT. Antivirulence genes: insights into pathogen evolution through gene loss. Infect Immun. 2012;80:4061–70 and. DOIPubMedGoogle Scholar
Figure
Table
Cite This ArticleTable of Contents – Volume 19, Number 8—August 2013
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Claire Jenkins, Microbiology Services, Health Protection Agency, 61 Colindale Ave, London NW9 5HT, UK
Top