Volume 20, Number 3—March 2014
Research
High-level Relatedness among Mycobacterium abscessus subsp. massiliense Strains from Widely Separated Outbreaks
Cite This Article
Citation for Media
Abstract
Three recently sequenced strains isolated from patients during an outbreak of Mycobacterium abscessus subsp. massiliense infections at a cystic fibrosis center in the United States were compared with 6 strains from an outbreak at a cystic fibrosis center in the United Kingdom and worldwide strains. Strains from the 2 cystic fibrosis outbreaks showed high-level relatedness with each other and major-level relatedness with strains that caused soft tissue infections during an epidemic in Brazil. We identified unique single-nucleotide polymorphisms in cystic fibrosis and soft tissue outbreak strains, separate single-nucleotide polymorphisms only in cystic fibrosis outbreak strains, and unique genomic traits for each subset of isolates. Our findings highlight the necessity of identifying M. abscessus to the subspecies level and screening all cystic fibrosis isolates for relatedness to these outbreak strains. We propose 2 diagnostic strategies that use partial sequencing of rpoB and secA1 genes and a multilocus sequence typing protocol.
Nontuberculous mycobacteria (NTM) and, in particular, the Mycobacterium abscessus group are recognized as emerging respiratory pathogens among patients with cystic fibrosis. Reports from the United States, France, and Israel have shown that the M. abscessus group accounts for a major proportion of NTM infections in patients with cystic fibrosis; prevalence rates range from 16% to 48% (1–3).
Previous studies have indicated great diversity within M. abscessus group strains among cystic fibrosis patients, suggesting independent acquisitions of NTM from the environment (2,4). However, suspicion of patient-to-patient transmission arose with the recent report of an outbreak of respiratory infection with M. abscessus subsp. massiliense at a cystic fibrosis center in Seattle, Washington, USA (5). The index case-patient and 4 additional patients all had multidrug-resistant isolates with resistance to amikacin and clarithromycin. All 5 strains were indistinguishable by repetitive unit sequence–based PCR patterns and pulsed-field gel electrophoresis analysis, which led to initiation of whole-genome sequencing. In a separate, recent study, whole-genome sequencing and epidemiologic analysis provided strong support for patient-to-patient transmission in 2 clustered outbreaks of M. abscessus subsp. massiliense at the Papworth Hospital Cystic Fibrosis Centre (Cambridge, UK) (6). Isolates from both clusters showed resistance to clarithromycin, and isolates from one of the clusters also had mutations conferring resistance to amikacin.
The availability of whole-genome sequences from different M. abscessus subsp. massiliense outbreaks, as well as unrelated strains, provides an unprecedented opportunity for multigenome comparisons. We conducted a genomic study of 3 recently sequenced strains from the Seattle cystic fibrosis outbreak, including the index strain, and compared them with representative strains from the Papworth cystic fibrosis outbreak, as well as with available strains from the United Kingdom, the United States, Brazil, South Korea, France, and Malaysia (Table 1). We found high-level relatedness among strains from the 2 geographically distant outbreaks in Seattle and Papworth. We also identified shared and unique genomic traits for strains from both cystic fibrosis outbreaks and for those from an outbreak of soft tissue infections in Brazil.
Sequence Analysis of Outbreak Strains
A subset of 6 isolates (2u, 12c, 14h, 19f, 20h, and 28c) representing the breadth of genomic diversity observed within the Papworth cystic fibrosis outbreak clusters 1 and 2 (6) were selected. Illumina sequencing reads from each of these isolates were assembled into sets of contigs by using Velvet software (21). These contigs were combined with draft genome sequences of the Seattle cystic fibrosis outbreak and available whole-genome sequences of M. abscessus subsp. massiliense (Table 1) and subjected to whole-genome multiple sequence alignments by using Mugsy software (22). Core segments of the alignment that are shared among all isolates included in the analysis were identified and concatenated by using Phylomark software (23). Concatenated nucleotide sequences, including single-nucleotide polymorphisms (SNPs), were then used for construction of a neighbor-joining phylogenetic tree by using MEGA software (24). The use of microbial samples and data was approved by the ethics committees at each of the institutions involved.
To replicate data from the Papworth cystic fibrosis outbreak clusters 1 and 2 (6) by using a similar approach, we mapped sequencing reads from the subset of 6 Papworth isolates, together with reads with from the 3 Seattle cystic fibrosis isolates and soft tissue strain CRM-0020 from Brazil (Table 1), onto the M. abscessus type strain ATCC 19977T reference genome by using BWA software (25). Variants, including SNPs, were called by using GATK software (26) and filtered for quality. The SNP panel was used for construction of a neighbor-joining phylogenetic tree by using MEGA software. The resulting tree replicated the topology of clusters 1 and 2 and showed that the Seattle isolates are most closely related to cluster 2.
PCR and In Silico PCR
Standard PCR and sequencing strategies were used to amplify and analyze partial sequences of the rpoB (723 bp) (27,28) and secA1 (465 bp) (29) genes. In addition, a multilocus sequence typing (MLST) scheme (29,30), including primers to 13 housekeeping genes (cya, gdhA, argH, glpK, gnd, murC, pgm, pknA, pta, pur, rpoB, hsp65, and secA1) was used to conduct electronic PCR on the panel of 20 M. abscessus subsp. massiliense genomes (Table 1). Published forward primers for cya and gdhA (30) did not amplify in silico for some M. abscessus subsp. massiliense strains; therefore, the following new primers conserved across the M. abscessus group were used: cya_F_new 5′-GCC TGC GTA AGG GTG ATG-3′ and gdhA_F_new 5′-GTG AAG CTC GCC GCC TGC-3′. Alleles from each gene were extracted and concatenated for each genome, the panel of concatenated sequences was aligned by using ClustalW software (31), and the core segments of the alignment were used for construction of a neighbor-joining phylogenetic tree by using MEGA software.
Phylogenetic Characteristics of Outbreak Strains
Figure 1

A core genome phylogenetic tree (Figure 1) showed a tight cluster of the 3 Seattle cystic fibrosis outbreak strains. The Seattle cystic fibrosis cluster was closely related to the 2 cystic fibrosis clusters described for the Papworth outbreak (6) and the Birmingham, UK, cystic fibrosis isolate 47J26 (9). Furthermore, the Seattle and Papworth cystic fibrosis outbreak strains showed some relatedness to strains CRM-0020 and GO-06 derived strains (known collectively as BRA-100) isolated during an epidemic of soft tissue infections in Brazil (32) and the M. abscessus subsp. massiliense M18 strain from Malaysia (10).
The cumulative size of core segments of Mugsy alignments provides information on relatedness among groups of strains compared. The core genome reduces in size as more genomes are added; an expected major decrease occurs after addition of more distant strains to the group. The average genome size of cystic fibrosis outbreak strains was 4.81 Mb for Seattle (n = 3) and 4.97 Mb for Papworth (n = 6). The Seattle and Papworth cystic fibrosis outbreak strains (n = 9) shared a core genome of 4,264,844 nt, which is almost unchanged by including the Birmingham cystic fibrosis strain 47J26 (n = 10; 4,264,127 nt). Addition of the soft tissue outbreak strain CRM-0020 from Brazil (n = 11) (32) decreased the core to 4,231,390 nt, and adding the related outbreak strain GO 06 from Brazil (n = 12) (8,33), led to an additional decrease in the core genome to 4,043,718 nt. As expected, including unrelated available clinical M. abscessus subsp. massiliense strains (n = 20, including M139 with ambiguous subspecies taxonomic assignment (12), (Table 1), reduced the core genome size to 3,869,950 nt. Further addition of M. abscessus subsp. abscessus (n = 2) and M. abscessus subsp. bolletii (n = 2) genomes (Table 1) reduced the core to 3,828,656 nt.
Genomes of Strains from Cystic Fibrosis and Soft Tissue Infection Outbreaks
The core genome of 10 strains representing the Papworth cystic fibrosis (n = 6), the Seattle cystic fibrosis (n = 3), and soft tissue CRM-0020 (n = 1) outbreaks comprised 4,231,390 nt. Strain GO 06 was excluded from the analysis because its genome harbors a large number of ambiguous nucleotides and an unusual hybrid appearance with fragments of M. abscessus subsp. massiliense and M. abscessus subsp. abscessus sequences. Strains 47J26 and M18, isolated from the sputum of a cystic fibrosis patient in Birmingham, UK, and a lymph node sample from a patient in Malaysia, respectively, were related to the outbreak strains (Figure 1). However, no information was available about any epidemiologic link between cystic fibrosis strain 47J26 to reported or unpublished outbreaks, and no clinical information was available about the patient from whom strain M18 was isolated. Therefore, both strains were excluded from the SNP analysis. Nevertheless, SNPs for these 3 strains at positions relevant to the outbreak strains are shown in the Technical Appendix.
Figure 2
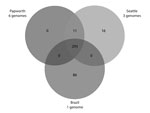
A total of 293 identical SNPs in the core segments of Mugsy alignments were shared by the 10 outbreak strains but were different in available M. abscessus subsp. massiliense strains not related to outbreaks (Figure 2; Technical Appendix). Of the 293 SNPs, 95 gave rise to nonsynonymous mutations in several genes, including virulence factors (mammalian cell entry and yrbE proteins), transcriptional regulators (TetR family), and lipid metabolism genes (Technical Appendix). Eleven SNPs were shared only by Papworth and Seattle cystic fibrosis outbreak strains (n = 9), including nonsynonymous mutations in the preprotein translocase secA1 and a putative lyase (Figure 2; Technical Appendix). Sixteen SNPs were shared only by the 3 Seattle cystic fibrosis outbreak strains, including nonsynonymous mutations in a mycobacterial large membrane protein (MmpL) family involved in lipid transport and virulence (34) and genes involved in amino acid and energy metabolism (Figure 2; Technical Appendix). Eighty-six SNPs were present only in strain CRM-0020 (soft tissue outbreak) from Brazil, including nonsynonymous mutations in an MmpL family protein; transcriptional regulators; and lipid, amino acid, and energy metabolism genes (Figure 2; Technical Appendix).
Having shown high-level relatedness among Papworth and Seattle cystic fibrosis outbreak strains and their relatedness to the soft tissue outbreak strains from Brazil, we also searched for genomic regions ≥200 nt outside the core genome that were specific to subsets of isolates. A single region of ≈11.5 kb was unique to the Papworth cystic fibrosis isolates (n = 6) and encompassed 2 conserved hypothetical proteins, 2 phage integrase family proteins, and an MmpL family protein. Alignment of the MmpL family protein with distinct MmpL proteins described above for the Seattle cystic fibrosis outbreak and the Brazil soft tissue outbreak showed diversity at several amino acid residues in all 3 proteins.
No region was unique to the Seattle cystic fibrosis isolates (n = 3). The soft tissue isolate CRM-0020 from Brazil harbored several large unique regions, including a previously described broad–host-range IncP-1β plasmid (35) and 3 regions (contigs) of 5 kb, 10.6 kb, and 79 kb of unknown origin encoding almost exclusively hypothetical proteins.
We also searched for polymorphisms associated with macrolide and aminoglycoside resistance. The Papworth cystic fibrosis and Seattle cytic fibrosis outbreak set of strains showed an A2058C/G mutation in 23S rRNA, which conferred macrolide resistance (36) (A2058G in strains 2u and 28c representative of Papworth cluster 2 and the Seattle strains). Strains 19f, 14h, 12c, and 28a, representative of Papworth cluster 1, and Seattle strains shared the A1408G mutations in 16S rRNA, which conferred aminoglycoside resistance (37). None of these mutations were found in the soft tissue outbreak strains CRM-0020 and GO 06 from Brazil or the M18 strain.
Diagnostic Tools for Identification of Outbreak Strains
In light of the possibility of a common ancestor and/or intercontinental transmission of strains, we identified SNPs in genes commonly used for identification of mycobacteria and an MLST scheme that could be used by clinical laboratories to assess relatedness of newly isolated strains to this global cluster. In the first approach, we retrieved rpoB sequences from the 6 genomes of representative strains of the Papworth cystic fibrosis outbreak and performed partial sequencing of the rpoB gene for selected isolates from the Seattle cystic fibrosis outbreak. We then compared these sequences with those of isolates from the outbreak in Brazil and unrelated clinical isolates comprising M. abscessus subsp. abscessus, massiliense, and bolletii, as well as other rapidly growing mycobacteria.
By using the rpoB gene MAB_3869c from the M. abscessus subsp. abscessus type strain as a reference (Table 2) described in the BRA-00 outbreak isolates from Brazil (32,33), we showed that Seattle (n = 4) and Papworth (n = 6) cystic fibrosis isolates carried the 2 rpoB SNPs (C→T at position 2569 and T→C at position 2760. However, none of the M. abscessus subsp. abscessus or subsp. bolletii or other rapidly growing mycobacterial isolates outside the M. abscessus group harbored this 2-SNP rpoB signature (Table 2). The second SNP (T→C substitution at position 2760) was present in several strains, but the combination of both rpoB SNPs (C→T at position 2569 and T→C at position 2760) was not present. Most of the 26 M. abscessus subsp. massiliense strains not related to outbreaks tested did not harbor this 2-SNP rpoB signature. However, 4 strains harbored this signature (Table 2) (29,38).
Multiple alignment of rpoB sequences among available M. abscessus subsp. massiliense genomes showed the absence of the 2-SNP rpoB signature in most strains. However, both SNPs were present in 1 strain not related to an outbreak (1S-151–0930) (Table 2).
Multiple alignment of secA1 sequences among available M. abscessus subsp. massiliense genomes showed a G→T substitution at position 820 (by using the secA1 gene MAB_3580c from the M. abscessus subsp. abscessus type strain) shared by the Papworth and Seattle cystic fibrosis outbreak strains but not by the soft tissue outbreak strains from Brazil or additional unrelated strains. Further analysis of secA1 sequences from 12 M. abscessus subsp. massiliense identified by multitarget sequencing and PCR-based typing (29,38) showed a G→T substitution at position 820 in 2 strains unrelated to the outbreak (Table 2). Those 2 strains were included among the 4 strains that had the 2-SNP rpoB signature. Although the SNPs described for rpoB and secA1 were not 100% specific markers for the outbreak strains, these SNPs could be used for first-level identification of newly isolated strains as possibly being related to cystic fibrosis clusters or soft tissue outbreak strains from Brazil to be confirmed by a second assay.
Figure 3

We also developed a simple MLST protocol that could be used as a second confirmatory assay. Alleles for each of 13 housekeeping genes (cya, gdhA, argH, glpK, gnd, murC, pgm, pknA, pta, pur, rpoB, hsp65, and secA1) were extracted and concatenated for each M. abscessus subsp. massiliense genome (Table 1), and the panel of concatenated sequences was used for construction of a neighbor-joining phylogenetic tree by using MEGA software. The Seattle and Papworth cystic fibrosis outbreak strains grouped together in the tree with cystic fibrosis strain 47J26 and isolate M18 from Malaysia (Figure 3). Thus, partial sequencing of rpoB and secA1 gens, followed by 13-target MLST analysis, could be used to rule out isolates as belonging to these 2 cystic fibrosis clusters.
The implications of this study are extensive. Currently, most experts recommend identifying isolates of M. abscessus to subspecies level (39). This report further corroborates these recommendations and places even greater pressure on clinical laboratories to fully identify M. abscessus subspecies massiliense.
Strains from the 2 cystic fibrosis outbreaks showed high-level relatedness (4,264,844 nt core genome alignment size, 11 shared unique SNPs) with each other and major-level relatedness (4,231,390 nt core genome alignment size) with soft tissue epidemic strains from Brazil. Genomic features shared between strains from all 3 outbreaks might make them more transmissible, whether from patient to patient (directly or indirectly as in cystic fibrosis outbreaks) or from a common source, as in soft tissue infections. However, the soft tissue strain from Brazil had the largest number of unique SNPs (86) not shared with either of the cystic fibrosis outbreak strains, harbored an IncP-1β plasmid, and did not show mutational resistance to amikacin or clarithromycin. We speculate that some of these specific genomic traits may be favorable for the successful establishment of epidemic soft tissue infections.
A previous study did not detect a common source or person-to-person transmission of the M. abscessus group among cystic fibrosis patients and suggested that it may not be necessary to segregate persons infected or colonized with M. abscessus from those who are not infected or colonized (40). Our findings emphasize the necessity of screening all isolates of M. abscessus subsp. massiliense recovered from patients with cystic fibrosis for relatedness to outbreak strains in an effort to prevent future outbreaks. Because of evidence supporting patient-to-patient transmission of multiple different respiratory tract organisms, the Infection Control Guidelines (currently in draft form for public comment) of the United States Cystic Fibrosis Foundation (CFF) (www.cff.org/LivingWithCF/Webcasts/ArchivedWebcasts/Germs/#Infection_Prevention_and_Control_Policy_Update) have been recently changed. Patients with cystic fibrosis are advised not to attend indoor meetings with other cystic fibrosis patients (CFF and Infection Prevention and Control Guidelines 2013). In addition, screening of all cystic fibrosis patients in the United States at least annually for mycobacteria is now recommended (CFF and Infection Prevention and Control Guidelines 2013) to enable early treatment if the organism is detected.
It remains unclear why intercontinental organisms are so closely related. One hypothesis is that direct patient contact led to transmission. The Seattle index case-patient traveled to British Columbia, Canada, before and after acquiring mycobacterial infection, to Oregon before mycobacterial infection, and to Atlanta, Georgia, and Bethesda, Maryland, after mycobacterial infection. However, the patient did not report any contact with other cystic fibrosis patients at these destinations. A second hypothesis is that the mycobacterial strain could have been carried by persons with cystic fibrosis who were clinically well. A third hypothesis is that there was an independent selection of M. abscessus subsp. massiliense clones in the cystic fibrosis airway milieu on both sides of the Atlantic Ocean toward potentially more transmissible lineages. Availability of additional whole-genome sequencing data tracking the global epidemiology of the M. abscessus group may help differentiate between these scenarios. In addition, this data will help delineate global clusters of M. abscessus subsp. massiliense strains with potentially higher transmissibility.
Addendum
Recent whole-genome data show deep genetic separation of 3 subspecies, ruling against grouping M. massiliense and M. bolletii under M. abscessus subsp. bolletii.
Dr Tettelin is an associate professor at the Institute for Genome Sciences, Department of Microbiology and Immunology, University of Maryland School of Medicine, Baltimore. His primary research interests are the use of comparative and functional genomics to understand bacterial diversity and virulence, study host-pathogen interactions, and identify vaccine candidates and drug targets to cure disease.
Acknowledgments
We thank Josephine Bryant, Dorothy Grogono, Julian Parkhill, and Andres Floto for their help and for providing sample identification and accession numbers for the Papworth outbreak isolates.
This study was supported in part by the National Institute of Allergy and Infectious Diseases (NIAID), the National Institutes of Health (NIH), the Department of Health and Human Services (contract no. HHSN272200900009C to C.M.F), and the Intramural Research Program (NIAID, NIH, Department of Health and Human Services). R.M.D, N.A.H, and M.S. were supported by the Amon G. Carter Foundation, the Colorado Bioscience Program, the Eppley Foundation, and the Boettcher Foundation. N.A.H. was supported by NIH Biomedical Informatics training grant 2T15LM009451-06. M.J. was supported by NIH/NIAID grant AI089718. B.B.-E. and R.J.W. were supported by Amon G. Carter Foundation.
References
- Levy I, Grisaru-Soen G, Lerner-Geva L, Kerem E, Blau H, Bentur L, Multicenter cross-sectional study of nontuberculous mycobacterial infections among cystic fibrosis patients, Israel. Emerg Infect Dis. 2008;14:378–84 . DOIPubMedGoogle Scholar
- Olivier KN, Weber DJ, Wallace RJ Jr, Faiz AR, Lee JH, Zhang Y, Nontuberculous mycobacteria. I: multicenter prevalence study in cystic fibrosis. Am J Respir Crit Care Med. 2003;167:828–34. DOIPubMedGoogle Scholar
- Roux AL, Catherinot E, Ripoll F, Soismier N, Macheras E, Ravilly S, Multicenter study of prevalence of nontuberculous mycobacteria in patients with cystic fibrosis in France. J Clin Microbiol. 2009;47:4124–8. DOIPubMedGoogle Scholar
- Sermet-Gaudelus I, Le Bourgeois M, Pierre-Audigier C, Offredo C, Guillemot D, Halley S, Mycobacterium abscessus and children with cystic fibrosis. Emerg Infect Dis. 2003;9:1587–91. DOIPubMedGoogle Scholar
- Aitken ML, Limaye A, Pottinger P, Whimbey E, Goss CH, Tonelli MR, Respiratory outbreak of Mycobacterium abscessus subspecies massiliense in a lung transplant and cystic fibrosis center. Am J Respir Crit Care Med. 2012;185:231–2. DOIPubMedGoogle Scholar
- Bryant JM, Grogono DM, Greaves D, Foweraker J, Roddick I, Inns T, Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: a retrospective cohort study. Lancet. 2013;381:1551–60 . DOIPubMedGoogle Scholar
- Davidson RM, Reynolds PR, Farias-Hesson E, Duarte RS, Jackson M, Strong M. Genome sequence of an epidemic isolate of Mycobacterium abscessus subsp. bolletii from Rio de Janeiro, Brazil. Genome Announc. 2013;1:e00617–13.
- Raiol T, Ribeiro GM, Maranhao AQ, Bocca AL, Silva-Pereira I, Junqueira-Kipnis AP, Complete genome sequence of Mycobacterium massiliense. J Bacteriol. 2012;194:5455. DOIPubMedGoogle Scholar
- Chan J, Halachev M, Yates E, Smith G, Pallen M. Whole-genome sequence of the emerging pathogen Mycobacterium abscessus strain 47J26. J Bacteriol. 2012;194:549. DOIPubMedGoogle Scholar
- Ngeow YF, Wong YL, Tan JL, Arumugam R, Wong GJ, Ong CS, Genome sequence of Mycobacterium massiliense M18, isolated from a lymph node biopsy specimen. J Bacteriol. 2012;194:4125. DOIPubMedGoogle Scholar
- Ngeow YF, Wong YL, Lokanathan N, Wong GJ, Ong CS, Ng KP, Genomic analysis of Mycobacterium massiliense strain M115, an isolate from human sputum. J Bacteriol. 2012;194:4786 and. DOIPubMedGoogle Scholar
- Ngeow YF, Wee WY, Wong YL, Tan JL, Ongi CS, Ng KP, Genomic analysis of Mycobacterium abscessus strain M139, which has an ambiguous subspecies taxonomic position. J Bacteriol. 2012;194:6002–3. DOIPubMedGoogle Scholar
- Choo SW, Wong YL, Tan JL, Ong CS, Wong GJ, Ng KP, Annotated genome sequence of Mycobacterium massiliense strain M154, belonging to the recently created taxon Mycobacterium abscessus subsp. bolletii comb. nov. J Bacteriol. 2012;194:4778. DOIPubMedGoogle Scholar
- Kim BJ, Kim BR, Hong SH, Seok SH, Kook YH. Complete genome sequence of Mycobacterium massiliense clinical strain Asan 50594, belonging to the type II genotype. Genome Announc. 2013;1:e00429–13.
- Tettelin H, Sampaio EP, Daugherty SC, Hine E, Riley DR, Sadzewicz L, Genomic insights into the emerging human pathogen Mycobacterium massiliense. J Bacteriol. 2012;194:5450 . DOIPubMedGoogle Scholar
- Adékambi T, Reynaud-Gaubert M, Greub G, Gevaudan MJ, La Scola B, Raoult D, Amoebal coculture of “Mycobacterium massiliense” sp. nov. from the sputum of a patient with hemoptoic pneumonia. J Clin Microbiol. 2004;42:5493–501 . DOIPubMedGoogle Scholar
- Pawlik A, Garnier G, Orgeur M, Tong P, Lohan A, Le Chevalier F, Identification and characterization of the genetic changes responsible for the characteristic smooth-to-rough morphotype alterations of clinically persistent Mycobacterium abscessus. Mol Microbiol. 2013;3:•••; Epub ahead of print and.PubMedGoogle Scholar
- Ripoll F, Pasek S, Schenowitz C, Dossat C, Barbe V, Rottman M, Non mycobacterial virulence genes in the genome of the emerging pathogen Mycobacterium abscessus. PLoS ONE. 2009;4:e5660. DOIPubMedGoogle Scholar
- Choi GE, Cho YJ, Koh WJ, Chun J, Cho SN, Shin SJ. Draft genome sequence of Mycobacterium abscessus subsp. bolletii BD(T). J Bacteriol. 2012;194:2756–7 . DOIPubMedGoogle Scholar
- Wong YL, Choo SW, Tan JL, Ong CS, Ng KP, Ngeow YF. Draft genome sequence of Mycobacterium bolletii strain M24, a rapidly growing mycobacterium of contentious taxonomic status. J Bacteriol. 2012;194:4475 . DOIPubMedGoogle Scholar
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9. DOIPubMedGoogle Scholar
- Angiuoli SV, Salzberg SL. Mugsy: fast multiple alignment of closely related whole genomes. Bioinformatics. 2011;27:334–42. DOIPubMedGoogle Scholar
- Sahl JW, Matalka MN, Rasko DA. Phylomark, a tool to identify conserved phylogenetic markers from whole-genome alignments. Appl Environ Microbiol. 2012;78:4884–92. DOIPubMedGoogle Scholar
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9. DOIPubMedGoogle Scholar
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. DOIPubMedGoogle Scholar
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. DOIPubMedGoogle Scholar
- Macheras E, Roux AL, Ripoll F, Sivadon-Tardy V, Gutierrez C, Gaillard JL, Inaccuracy of single-target sequencing for discriminating species of the Mycobacterium abscessus group. J Clin Microbiol. 2009;47:2596–600. DOIPubMedGoogle Scholar
- Adékambi T, Colson P, Drancourt M. rpoB-based identification of nonpigmented and late-pigmenting rapidly growing mycobacteria. J Clin Microbiol. 2003;41:5699–708. DOIPubMedGoogle Scholar
- Zelazny AM, Root JM, Shea YR, Colombo RE, Shamputa IC, Stock F, Cohort study of molecular identification and typing of Mycobacterium abscessus, Mycobacterium massiliense, and Mycobacterium bolletii. J Clin Microbiol. 2009;47:1985–95. DOIPubMedGoogle Scholar
- Macheras E, Roux AL, Bastian S, Leao SC, Palaci M, Sivadon-Tardy V, Multilocus sequence analysis and rpoB sequencing of Mycobacterium abscessus (sensu lato) strains. J Clin Microbiol. 2011;49:491–9. DOIPubMedGoogle Scholar
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80. DOIPubMedGoogle Scholar
- Duarte RS, Lourenco MC, Fonseca Lde S, Leao SC, Amorim Ede L, Rocha IL, Epidemic of postsurgical infections caused by Mycobacterium massiliense. J Clin Microbiol. 2009;47:2149–55. DOIPubMedGoogle Scholar
- Leão SC, Viana-Niero C, Matsumoto CK, Lima KV, Lopes ML, Palaci M, Epidemic of surgical-site infections by a single clone of rapidly growing mycobacteria in Brazil. Future Microbiol. 2010;5:971–80. DOIPubMedGoogle Scholar
- Varela C, Rittmann D, Singh A, Krumbach K, Bhatt K, Eggeling L, MmpL genes are associated with mycolic acid metabolism in mycobacteria and corynebacteria. Chem Biol. 2012;19:498–506. DOIPubMedGoogle Scholar
- Leão SC, Matsumoto CK, Carneiro A, Ramos RT, Nogueira CL, Lima JD Jr, The detection and sequencing of a broad-host-range conjugative IncP-1beta plasmid in an epidemic strain of Mycobacterium abscessus subsp. bolletii. PLoS ONE. 2013;8:e60746. DOIPubMedGoogle Scholar
- Wallace RJ Jr, Meier A, Brown BA, Zhang Y, Sander P, Onyi GO, Genetic basis for clarithromycin resistance among isolates of Mycobacterium chelonae and Mycobacterium abscessus. Antimicrob Agents Chemother. 1996;40:1676–81 .PubMedGoogle Scholar
- Prammananan T, Sander P, Brown BA, Frischkorn K, Onyi GO, Zhang Y, A single 16S ribosomal RNA substitution is responsible for resistance to amikacin and other 2-deoxystreptamine aminoglycosides in Mycobacterium abscessus and Mycobacterium chelonae. J Infect Dis. 1998;177:1573–81. DOIPubMedGoogle Scholar
- Shallom SJ, Gardina PJ, Myers TG, Sebastian Y, Conville P, Calhoun LB, New rapid scheme for distinguishing the subspecies of the Mycobacterium abscessus group and identification of Mycobacterium massiliense with inducible clarithromycin resistance. J Clin Microbiol. 2013;51:2943–9. DOIPubMedGoogle Scholar
- Koh WJ, Jeon K, Lee NY, Kim BJ, Kook YH, Lee SH, Clinical significance of differentiation of Mycobacterium massiliense from Mycobacterium abscessus. Am J Respir Crit Care Med. 2011;183:405–10. DOIPubMedGoogle Scholar
- Bange FC, Brown BA, Smaczny C, Wallace RJ Jr, Bottger EC. Lack of transmission of Mycobacterium abscessus among patients with cystic fibrosis attending a single clinic. Clin Infect Dis. 2001;32:1648–50. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 20, Number 3—March 2014
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Adrian M. Zelazny, Department of Laboratory Medicine, National Institutes of Health Clinical Center, 10 Center Dr, Bldg 10-2C385, Bethesda, MD 20892-1508, USAAdrian M. Zelazny, Department of Laboratory Medicine, National Institutes of Health Clinical Center, 10 Center Dr, Bldg 10-2C385, Bethesda, MD 20892-1508, USA
Top