Volume 22, Number 12—December 2016
Letter
New Hepatitis E Virus Genotype in Bactrian Camels, Xinjiang, China, 2013
Figure
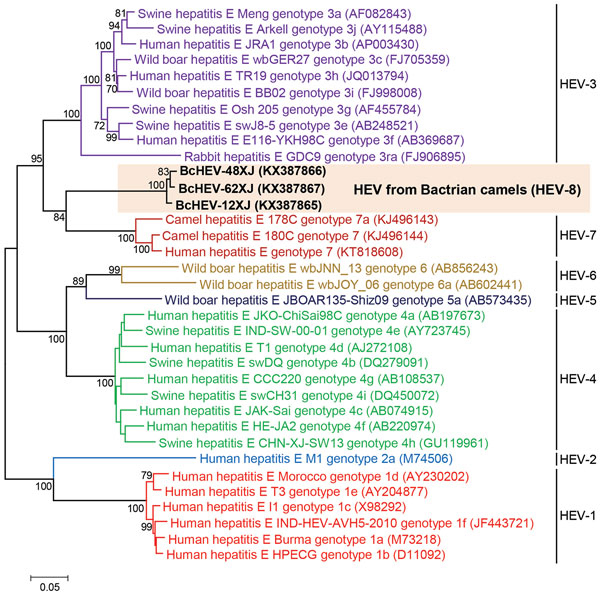
Figure. Phylogenetic analyses of the proteins of concatenated ORF1/ORF2, excluding the hypervariable region, of Bactrian camel hepatitis E virus (HEV) and other HEV genotypes (HEV1–HEV7) within the species Orthohepevirus A (family Hepeviridae). The tree was constructed using the maximum-likelihood method using the Jones–Taylor–Thornton substitution model with invariant sites and gamma distributed rate variation. The analysis included 2,282 amino acid positions (aa residues 1–706 and 789–2409, numbered with reference to GenBank sequence M73218). Bold indicates the 3 strains of BcHEV with complete genomes sequenced in this study. GenBank accession numbers are shown in parentheses. Scale bar indicates the estimated number of substitutions per 20 aa. ORF, open-reading frame.
1These authors contributed equally to this article.